
食用植物油中甾醇总含量测定方法的优化
2022-10-26 04:43:18翟孟婷林泽峰张江旭王翔宇
中国粮油学报 2022年9期
翟孟婷, 谢 亮, 林泽峰, 张江旭, 王翔宇,2, 惠 菊,2
(中粮营养健康研究院有限公司,营养健康与食品安全北京市重点实验室, 老年营养食品研究北京市工程实验室1,北京 102209) (江苏省现代粮食流通与安全协同创新中心2,南京 210023)
甾醇是天然甾族化合物中的一类物质,按照来源可分为动物甾醇、植物甾醇和菌甾醇。植物甾醇是一种天然活性物质,通常为无色、无味、不溶于水、易溶于有机物,结构上与胆固醇相似[1]。植物甾醇主要来源于植物类食物,且大部分存在于油料种子中[2]。食用植物油中含有多种甾醇,甾醇含量相对较多的为稻米油、玉米油、葵花籽油、菜籽油,主要甾醇种类为菜籽甾醇、菜油甾醇、豆甾醇、β-谷甾醇。植物甾醇应用范围很广,有助于降低血清总胆固醇、低密度脂蛋白胆固醇以及血清甘油三酯的浓度,添加到化妆品中可提高乳化稳定性,添加到饲料中可以提高禽畜的增重率[1]。目前食用植物油中甾醇提取方法主要为碱溶液加热皂化提取结合液液萃取法[3,4],薄层色谱法[5]或固相萃取法[6,7]净化提纯后上机测定。文献报道的甾醇分析方法主要有气相色谱法[6-8],气相色谱质谱法[9,10],液相色谱法[11,12],液相-气相色谱联用法[13,14]。气相色谱法的分离度和灵敏度相对较好,设备价格相对较低且对操作人员要求不高等优势,国家标准中使用碱液皂化-氧化铝柱除杂-薄层板分离提纯结合气相色谱定量国家标准方法具有适用性广,甾醇净化率高的优势,但因操作过程繁琐且耗时长,无法批量处理。气相色谱质谱法灵敏度和抗干扰力较强,是近几年甾醇分析使用较多的方法。液相色谱对甾醇的分离稳定性相对较差,有文献报道液相-气相色谱联用法,液相色谱对样品进行分离,收集洗脱结合气相色谱进行测定完成15种甾醇组分的分析,样品前处理相对简单易操作,但分析时间较长对设备的要求比较高,不能满足快速测定需求[13]。
本研究选择前处理方法简单且设备灵敏度和分离度能满足要求的检测方法,在改进国家标准方法基础上,优化样品前处理过程,人员实验技能要求较低同时满足油脂工厂快速测定甾醇的要求,结合气相色谱完成甾醇化合物分析。
1 材料与方法
1.1 实验材料与试剂
甾醇标品∶胆固醇99%,菜籽甾醇,豆甾醇,菜油甾醇,β-谷甾醇。
衍生化试剂(BSTFA∶TMCS=99∶1),正己烷,色谱纯; 95%乙醇,分析纯;氢氧化钾, 无水硫酸钠,15 mL塑料离心管。
1.2 仪器与设备
7890B气相色谱-FID检测器,HP-5色谱柱(30 m×0.32 mm×0.25 μm),TTL-DC11氮吹仪,ST-16R离心机,水浴锅,涡旋混合器,ME-204天平。
1.3 方法
1.3.1 标准溶液配制
胆固醇标准溶液:准确称量100 mg胆固醇标品(准确到0.100 mg),用正己烷溶解,于25 mL容量瓶中定容。将溶液转移至棕色试剂瓶中,密封后,置于-20 ℃保存1周。
菜籽甾醇、豆甾醇和β-谷甾醇标准溶液(1.00 mg/mL):分别准确称量5 mg甾醇标品(准确到0.100 mg),用正己烷溶解,于5 mL容量瓶中定容。将溶液转移至棕色试剂瓶中,密封后,在-20 ℃保存3个月。
菜油甾醇标准溶液(0.10 mg/mL):称量1 mg菜油甾醇标品,取少量正己烷于标准品瓶中重复3次,将标准品溶解后转移至进样瓶中,密封后,在-20 ℃保存3个月。
甾醇工作溶液:量取1 mL菜籽甾醇、菜油甾醇、豆甾醇、β-谷甾醇标准溶液于进样瓶中衍生化后,气相色谱定性分析。
1.3.2 样品处理
称取200 mg样品于15 mL离心管中,依次加入500 μL内标溶液,2 mol/mL氢氧化钾-乙醇溶液1 mL,涡旋1 min,混匀,超声3 min,取出后加入4 mL KOH-乙醇溶液,涡旋1 min后放入60 ℃水浴锅中加热1 h,取出后冷却。加入2 mL去离子水,5 mL正己烷,涡旋后4 000 r/min离心10 min,将上层溶液转移至另一15 mL离心管中,重复提取2次,加入1 g无水硫酸钠。将提取液氮吹至2 mL,离心后过0.22 μm有机滤膜,转移至进样瓶中,氮吹至干,再加入400 μL衍生化试剂,60 ℃水浴加热1 h,上机分析。
1.3.3 气相色谱条件
HP-5毛细管柱(30 m×0.32 mm×0.25 μm),载气:高纯氮气,纯度99.999%;恒压,16.349 psi;分流比:20∶1;进样口温度:320 ℃;检测器温度:320 ℃;程序升温:初始温度240 ℃,保持1 min,以4℃/min的速率从240 ℃升温到255 ℃,保持35 min;进样量:1.0 μL。
1.3.4 结果计算
本方法的甾醇总量计算方法,参考GB/T 25223—2010,甾醇总量的计算从菜籽甾醇到Δ7-燕麦甾烯醇所有甾醇的峰。
式中:S为试样中甾醇总量,以100 g油中含有的质量表示/mg/100 g;mB为胆固醇的质量/mg;Σ(A)为单体甾醇峰面积的和;AB为胆固醇内标峰面积;mT为试样的质量/g。
1.3.5 数据分析
本文样品测定方法的甾醇总量计算方法参考GB/T 25223—2010,实验数据用Excel表分析,用Origin软件作图。
2 结果与分析
2.1 前处理条件优化
2.1.1 碱液浓度
称取200 mg样品按照分别添加0.5、1、2、2.5 mol/mL浓度碱液5 mL,进行样品前处理后上机分析。探究碱液浓度对甾醇含量的影响,结果见图1。碱液浓度增加有助于皂化物的提取,碱液浓度2.5 mol/mL 和2 mol/mL时,甾醇总量值上升为最大值,碱液浓度过高时,溶液黏度略大,不利于萃取,因此浓度为2 mol/mL最优。
2.1.2 提取溶剂用量
因乙醚与乙醇容易发生乳化反应,回收率略低于正己烷[15]。本实验选择正己烷为提取溶剂,并对正己烷的用量进行探究。核桃油中豆甾醇本底值较低为14.25 mg/kg。称取核桃油200 mg,添加豆甾醇标品200 μg,按照1.3.2进行前处理,选取正己烷4、8、10、12 mL对皂化液进行2次萃取,上机分析。豆甾醇回收率随着正己烷用量增加而增高在10 mL后开始。在本实验条件下,正己烷用量4 mL时,有乳化现象分层不明显。当正己烷用量8、10 mL时回收率最高,分别为99.72%、100.21%,过多的提取液在氮吹浓缩耗时较长因此本实验选择使用正己烷8 mL,结果见图1。

图1 碱液浓度以及正己烷用量对甾醇的影响
2.1.3 前处理条件对结果影响探究
薄层色谱分离可以将不皂化物中的甾醇与烃类、三萜烯醇和甲基甾醇等分开,其分离效果影响甾醇结果的准确性[16]。本研究省略薄层色谱分析步骤,具有快速测定批量处理的优势,探究本方法与GB/T 25223—2010测定甾醇总量结果差异,选取2个玉米毛油结果进行比较,结果见表1。对2个样品的2个总体均值之差检验,计算的2个P值,均大于P=0.05,因此2种检测方法差异性不显著。
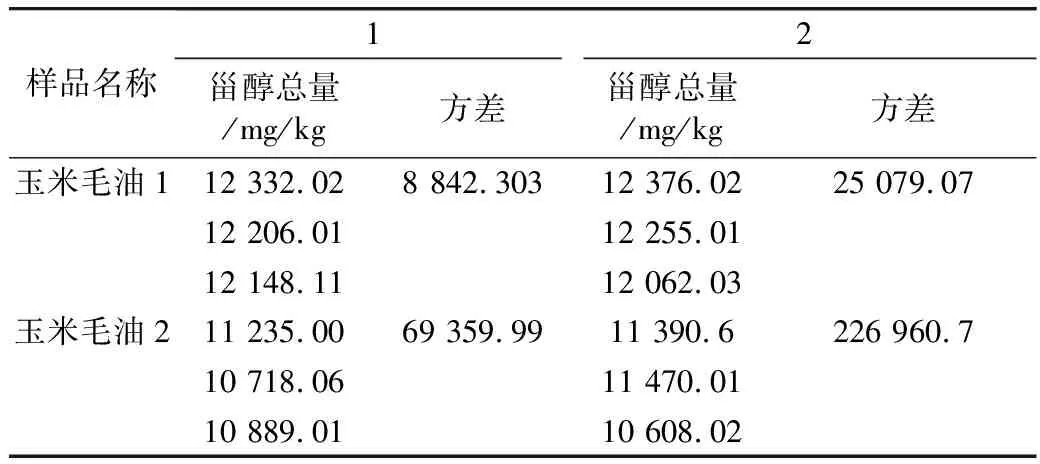
表1 2种方法测定玉米油中甾醇总量表
2.2 色谱分离
本实验使用Aglient HP-5非极性毛细管柱,具有良好的惰性和热稳定性,可以较短时间内实现甾醇完全分离。菜籽甾醇、菜油甾醇、豆甾醇和β-谷甾醇标准品色谱图,如图2所示。
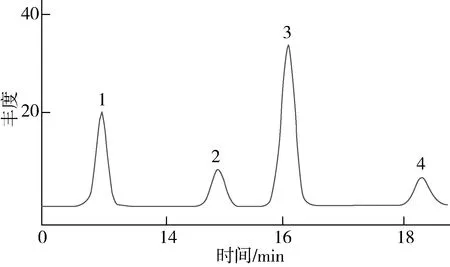
注:1 菜籽甾醇;2 菜油甾醇;3 豆甾醇;4 β-谷甾醇。图2 4种甾醇标品色谱图
2.3 甾醇鉴定
国家标准方法中将氢气作为载气,使用SE-54色谱柱进行分离给出甾醇的相对保留时间,因氢气在实验室安全风险系数较高,对气体储存要求极高,不适用于一般实验室。所以本方法使用氮气作为载气, HP-5色谱柱分离测定相对保留时间(RRT)鉴定植物油中的甾醇种类,以待测甾醇保留时间除以胆固醇的保留时间(RT),表2列出了在本实验条件下12种甾醇的相对保留时间。
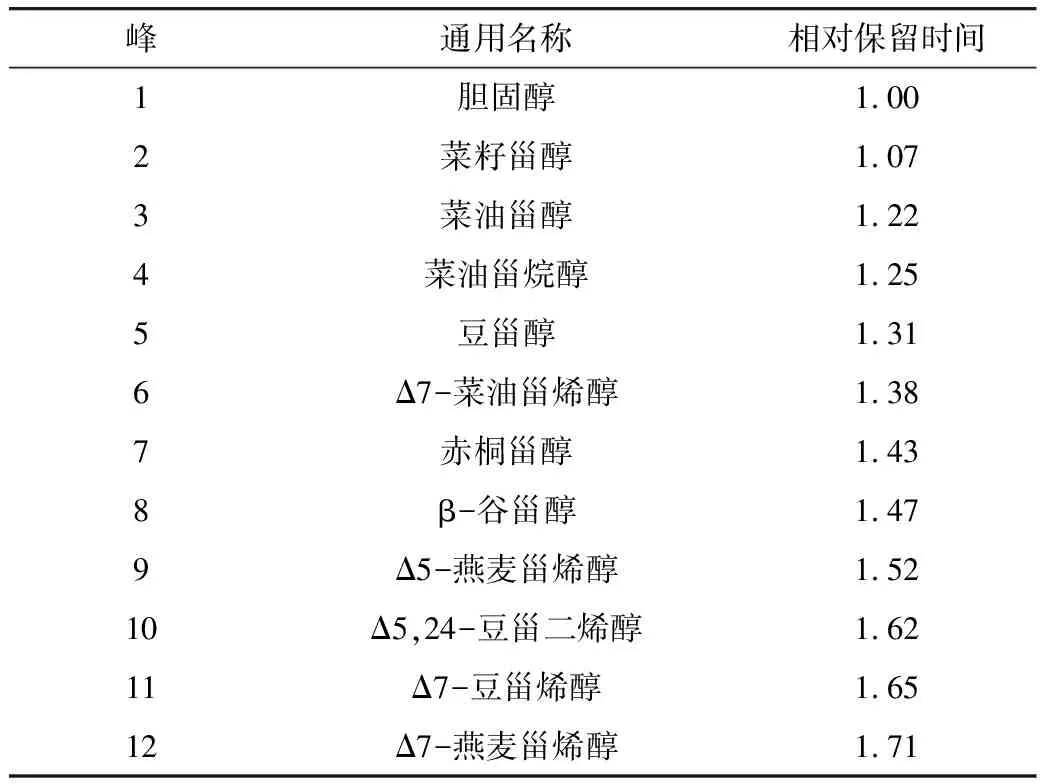
表2 甾醇相对保留时间表
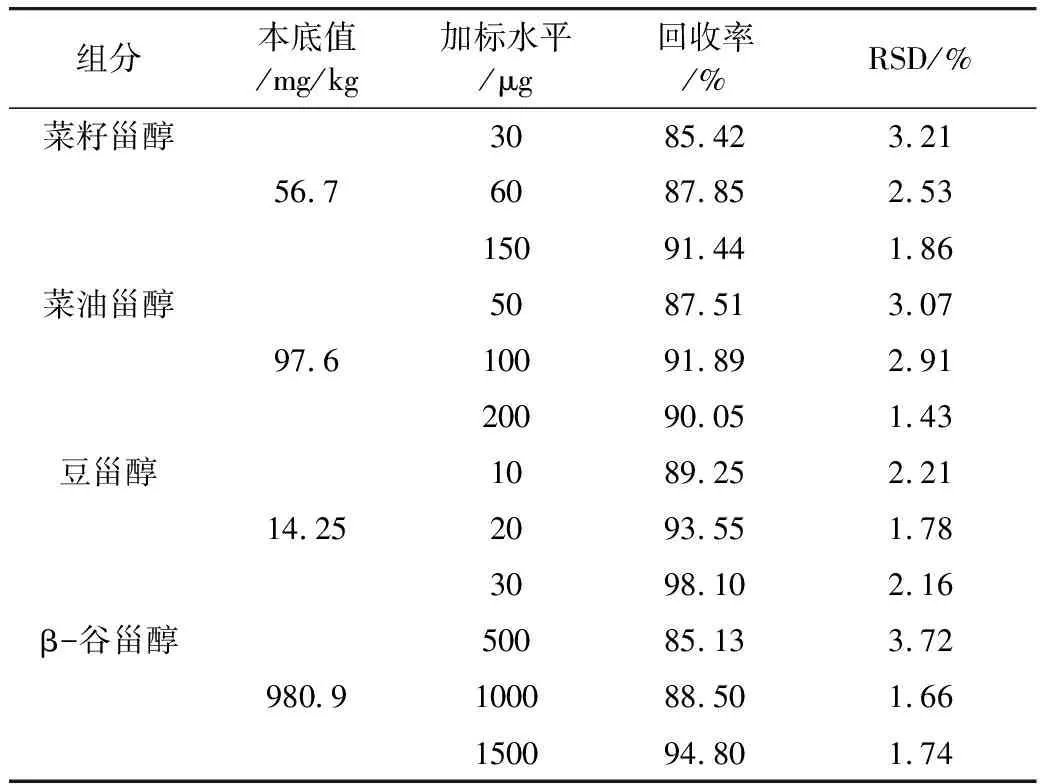
表3 方法添加回收率
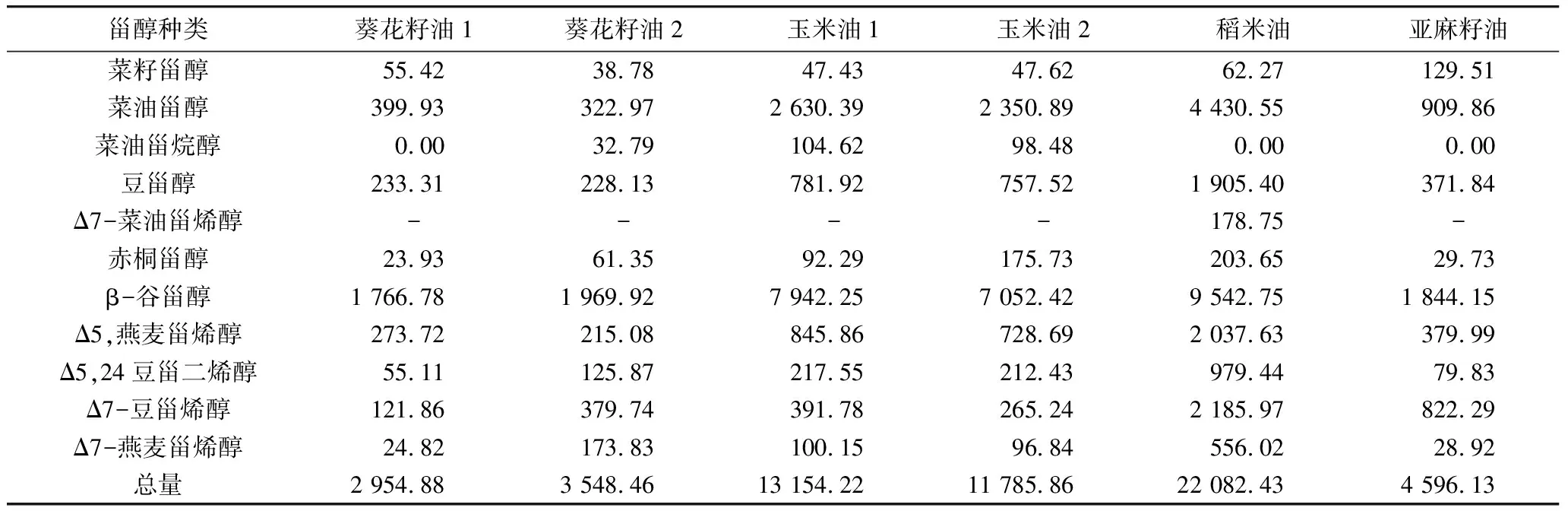
表4 4种食用植物油中甾醇含量测定值/mg/kg

表5 6种食用植物油中甾醇含量测定值/mg/kg
2.4 回收率和精密度
考虑样品本底值的基础上,对核桃油进行4类甾醇3种浓度水平进行3次添加回收实验,其中菜籽甾醇回收率为85.42%~91.44%,菜油甾醇回收率为87.51%~91.89%,豆甾醇回收率为89.25%~98.10%,β-谷甾醇回收率为85.13%~94.80%,该方法的相对标准偏差为1.43%~3.72%。β-谷甾醇低浓度添加回收率略低,可能与样品本底值较高有关,详见表3。
2.5 方法检出限和定量限
本文保证结果的稳定性和准确性,样品加入内标定量。标品储备液逐级稀释,以3倍信噪比(S/N)计算方法的检出限(LOD)为0.11 mg/kg,10倍信噪比计算定量限(LOQ)为0.25 mg/kg。
2.6 不同植物油中的甾醇含量
食用植物油中主要以菜油甾醇、豆甾醇、β-谷甾醇为主。由表4、表5可知,菜油甾醇在7种油中含量较高,β-谷甾醇在10种油中含量最高,豆甾烯醇在山茶油、胡麻籽油和芝麻油中含量较高,菜油甾烯醇仅在稻米油中检出。花生油、葵花籽油,山茶油和胡麻籽油甾醇总量在2 839~4 798 mg/kg范围内,核桃油甾醇总量最低为1 794.22 mg/kg。稻米油甾醇总量最高为22 082mg/kg,其次为玉米油11 000~13 000 mg/kg,这2种油的Δ5-燕麦甾烯醇和Δ5,24-豆甾烯醇含量高于其他品类油。
3 结论
本文建立快速测定油脂中甾醇方法,在国家标准基础上进行优化,样品氢氧化钾乙醇溶液加热皂化后少量正己烷提取,氮吹浓缩衍生反应后,利用气相色谱进行12种甾醇分析。本方法简便高效,适合高通量处理且减少有机试剂使用量,节省实验成本同时对实验人员友好健康,满足油脂工厂快速测定甾醇的需求。
猜你喜欢
CHINA TODAY(2022年4期)2022-11-22 12:43:48
花卉(2021年17期)2021-09-15 08:51:16
中国粮油学报(2018年12期)2018-03-19 05:40:44
农产品市场周刊(2018年37期)2018-01-27 12:17:29
中国粮油学报(2017年2期)2017-08-07 03:37:17
北方牧业(2016年6期)2016-12-17 14:04:49
中国粮油学报(2016年5期)2016-01-23 02:44:53
小天使·四年级语数英综合(2015年10期)2015-10-14 18:01:40
伴侣(2015年8期)2015-08-11 02:30:32
中国粮油学报(2015年5期)2015-02-06 01:47:23
