
乙基纤维素微胶囊化膨化小麦胚芽粉品质研究
2022-10-26 03:44王旭成周柏玲孟婷婷许效群
中国粮油学报 2022年9期
王旭成, 周柏玲, 孟婷婷, 田 歌, 许效群
(山西农业大学食品科学与工程学院1,太谷 030801) (山西农业大学山西功能食品研究院2,太原 030006)
小麦是一种禾本科单子叶植物,完整的小麦籽粒由皮层、胚芽和胚乳三部分组成,其中胚芽一般占小麦籽粒总质量的1.4%~3.9%[1],富含优质的蛋白质、脂肪、糖类、多种维生素和矿物质以及一些人体所需的生理活性成分[2-4]。由于籽粒中胚芽与胚乳结合不紧密,在小麦加工过程中往往自然脱落,成为副产物或其他加工的原料,且由于其中含有不饱和脂肪酸的缘故,若保存不当,会造成胚芽资源的极大浪费[5]。
微胶囊化作为一种日渐成熟的加工技术,其对微胶囊化目标有着良好的保护、改变形态、缓释、掩盖不良气味等作用,被广泛应用于日用品、医药、畜牧养殖、食品、化学材料等领域[6-8]。制备微胶囊的方法较多,其中空气悬浮法(流化床包衣法)是通过物理机械方法制备微囊的主要方式。空气悬浮法制备微胶囊是将固体颗粒作为芯材,高分子聚合物作为壁材,将芯材放置于流化床中,使其在喷射气流的作用下快速规则运转,当芯材通过包衣区域时,壁材溶液在高气压作用下呈雾化状均匀喷出,附着在芯材的表面,随着有机溶剂的挥发,壁材在芯材表面形成多层的高分子膜,以此达到包埋效果[9]。该方法可以包埋固体颗粒,便于工厂流水线大规模批量作业,泛用性更强[10,11]。乙基纤维素(EC)具有良好的成膜性和疏水性,热稳定性较好[12],在低温下仍能保持挠曲性,有极强的抗生物性能,无毒,是最常用的缓控释包衣材料之一,被医药领域广泛使用[13-15]。
目前关于微胶囊化膨化小麦胚芽粉的研究鲜有报道。食品工业领域主流的微胶囊化技术为喷雾干燥法,这一方法对芯材的溶解度有较高的要求,且成本较高。本实验采用部分可溶的膨化小麦胚芽粉为芯材,不同浓度乙基纤维素-乙醇溶液为壁材溶液,使用空气悬浮法,制得对应的微胶囊样品,通过对比微胶囊样品的相关指标,即含水量、溶解度、容积密度、流动性、包含率、粒径分布、感官评价、表面形态结构和加速储藏条件下的脂肪酸值的变化,对微胶囊样品的产品品质、抗氧化性和成本进行综合考虑,选出乙基纤维素-乙醇溶液微胶囊化膨化小麦胚芽粉的最适浓度,为小麦胚芽深加工提供可行思路和参考方向。
1 材料与方法
1.1 材料与仪器
1.1.1 实验材料
小麦胚芽挤压膨化产物(以小麦胚芽粉为原料,使用双螺杆挤压膨化机,物料水分为25%,末端温度为120 ℃,螺杆转速为50 r/min,制得相应膨化产物)。
1.1.2 主要试剂
乙基纤维素、海藻酸钠、乙醇、氢氧化钾、酚酞,分析纯。
1.1.3 实验仪器与设备
2200型不锈钢磨粉机,FDV超微粉碎机,101-OA电热鼓风干燥箱,DZKW-4电热恒温水浴锅,LBF-5型旋转流化床制粒包衣机,LXJ-IIB低速大容量离心机,BT-2001激光粒度分布仪,BT-901干法分散进样系统,Helios G4 UC等线聚焦离子束-电子束双束电镜。
1.2 方法
1.2.1 制备膨化胚芽粉
使用不锈钢磨粉机将小麦胚芽挤压膨化产物初步粉碎,转速为1 500 r/min,再使用超微粉碎机将其进一步粉碎,转速为22 000 r/min,制得膨化胚芽粉。
1.2.2 制备膨化胚芽微粒
由于经过超微粉碎的样品中存在粒径过大和过小的颗粒,其会影响样品微胶囊化的效果,谢中国等[16]通过在微胶囊化前的大黄鱼仔稚幼体饲料中添加海藻酸钠的方式,使得饲料颗粒粒径均匀度提高,有效提升微胶囊化后产品质量。向制备样品中加入制备样品总质量1.00%的海藻酸钠,通过人工翻转搅拌的方式混匀,再加入制备样品总质量8.00%的蒸馏水,再次进行混匀,将混匀的样品过40目网筛,用自封袋收集筛下样品,收集物即制得的膨化胚芽微粒。
1.2.3 制备不同浓度EC溶液
配制0.00%、2.00%、2.75%、3.50%、4.25%和5.00%质量浓度的EC溶液,分别称取对应质量EC,采用无水乙醇为溶剂,使用恒温水浴锅于50 ℃加热溶解,备用。
1.2.4 制备不同浓度EC溶液微胶囊样品
本实验采用膨化胚芽微粒为芯材,不同浓度EC溶液为壁材溶液,使用空气悬浮法,包衣方式为底喷,气源压强0.45 MPa,气密压强0.3 MPa,壁材流量2 mL/min,进风温度50 ℃,出风温度30 ℃,床温40 ℃,包衣时间120 min[17-20]。将制备好的微胶囊样品装入自封袋中,于4 ℃环境下保存备用。
1.2.5 含水量的测定
称取5 g微胶囊样品置于已干燥至恒重的铝盒中,于105 ℃烘箱干燥5 h,冷却称重。再次重复操作,直至2次质量差小于1 mg,记录此时样品与铝盒的总质量[21]。按式(1)计算微胶囊样品的含水量(X)。
(1)
式中:m0为铝盒质量/g;m1为烘干前样品与铝盒的总质量/g;m2为烘干后样品与铝盒的总质量/g。
1.2.6 容积密度测定
将微胶囊样品倒入10 mL已干燥至恒重的刻度试管中,称重。按式(2)计算微胶囊样品的容积密度(ρ)[22]。
(2)
式中:m0为刻度试管质量/g;m1为样品与刻度试管的总质量/g;V为刻度试管体积/mL。
1.2.7 溶解度测定
称取5 g的微胶囊样品,放置于50 mL的小烧杯中,加入40 mL水温在25~30 ℃之间的蒸馏水,少量多次溶解后,转移到50 mL离心管,在4 000 r/min条件下离心10 min,弃去上清液。再次重复操作2~3次。将剩余沉淀用蒸馏水洗出至已干燥至恒重的称量皿中,蒸干水分,冷却称质量[23]。按式(3)计算微胶囊样品的溶解度(Y)。
(3)
式中:X为样品含水量/%;m为样品质量/g;m1为称量皿质量/g;m2为烘干后称量皿与沉淀的总质量/g。
1.2.8 流动性测定
于水平桌面放置一块平板,在其上方固定一个玻璃漏斗,精确称取10 g微胶囊样品置于漏斗中,让样品自由通过漏斗,于平板上自然堆积,测定粉堆的高度与覆盖半径[24]。按式(4)计算微胶囊样品的休止角(θ)。
θ=arctan(h/r)
(4)
式中:θ为样品休止角/(°);h为样品粉堆的高度/cm;r为样品粉堆的覆盖半径/cm。
1.2.9 包含率测定
称取5 g的微胶囊样品,按1.2.4中方法测定其含水量(X)。按式(5)计算微胶囊样品的干物质含量(Z)。
Z=1-X
(5)
式中:X为样品含水量/%。
芯材干物质的含量与微胶囊样品含量的比值为包含率。按式(6)计算微胶囊样品的包含率(A)。
(6)
式中:Z为样品干物质质量分数/%;Z0为芯材干物质质量分数/%。
1.2.10 样品粒径分布的测定
使用激光粒度分布仪,采用干法测定体系,对微胶囊样品进行粒径分布的测定[25]。
1.2.11 感官评价
邀请10名经过专业培训的感官评价人员组成感官品评小组,对刚制得的微胶囊样品的色泽、组织结构、质地、气味进行评定并打分,满分为100分,90分以上为优秀,80~90分为良好,60~80分为合格,60分以下为差,评分标准细则见表1。
1.2.12 观察样品表面形态结构
用导电双面胶将样品固定在金属样品平台上,在真空中喷涂钯金后,置于双束电镜中以3.00 kV电子束观察微胶囊的表面形态结构,并选择样品分布较均匀的视野进行拍照[26]。
1.2.13 加速储藏条件下样品脂肪酸值变化的测定
结合姜平等[27]采用的加速储藏方法,将微胶囊样品置于60 ℃的烘箱中储藏,储藏期为30 d,每隔3 d取样,按GB/T 15684—2015测定样品的脂肪酸值。
1.2.14 统计学分析
所有实验至少重复3次,所得数据用“平均值±标准偏差”表示,采用Origin 8.0软件绘图,采用SPSS 18比较均值中的单因子方差分析进行统计分析,采用Duncan进行两两比较,显著性水平为P<0.05。
2 结果与分析
2.1 样品的基础指标
由表2可知,不同浓度EC溶液制得的样品基础指标存在较大差距。就样品含水量和溶解度而言,含水量跨度为1.98%,溶解度跨度为5.02%,随着EC溶液浓度的升高,样品含水量下降,样品溶解度下降。由于EC是一种不溶于水的高分子聚合物,有较强的疏水性[28],当其覆盖于样品颗粒表面时,会影响样品吸水性和在水中的溶解性,使得样品含水量下降和在水中的溶解度下降。就样品密度和休止角而言,密度跨度为7.50 g/100 mL,休止角跨度为17.6°,随着EC溶液浓度的升高,样品密度下降,休止角下降,即流动性上升。休止角低于30°,样品流动性良好,休止角在30°~45°,样品流动性较好,休止角大于45°,样品的流动性较差[29],故4.25%和5.00%EC溶液微胶囊化样品流动性良好,2.75%、3.50%、4.25%EC溶液微胶囊化样品流动性较好,0%EC溶液微胶囊化样品流动性较差,由于EC溶液具有较高黏度[30],随着EC浓度的升高,其黏度也随之升高,在包埋过程中样品颗粒黏合的问题加剧,导致样品中黏合物比例提高,影响样品颗粒粒径以及均匀度,造成样品颗粒粒径提高以及均匀度下降,使得样品密度下降和流动性上升。就样品包含率而言,包含跨度为2.09%,随着EC溶液浓度的升高,包含率下降,由于EC溶液浓度的升高,单位质量内干物质比例下降,即芯材比例下降,使得样品包含率下降。

表2 胚芽系列样品基础指标
2.2 样品粒径分布
由图1a可知,0.00%EC溶液微胶囊化样品图谱只存在1个峰,即样品区间分布主要集中在20.80~24.34 μm处,符合正态分布,说明样品均匀度较为良好。2.00%~5.00%EC溶液微胶囊化样品图谱都存在2个峰,不符合正态分布,说明样品均匀度较差,符合样品密度和流动性中得出的结论,即EC溶液浓度升高,均匀度下降;样品最高峰在200.0 μm处,随着EC溶液的升高,样品在200.0 μm处的峰值升高,说明EC溶液微胶囊化胚芽微粉,会将粒径较小的颗粒聚合包埋至粒径200.0 μm左右,且随着EC溶液浓度升高,样品中200.0 μm左右的颗粒占比越大。由图1b可知,0%EC溶液微胶囊化样品累积量在42.16~45.61 μm处超过90%,2.00%EC溶液微胶囊化样品的累积量在67.53~73.05 μm处超过90%,5.00%EC溶液微胶囊化样品的累积量在200.0~300.0 μm处超过90%,随着EC溶液的升高,样品累积量超过90%所在的粒径值越大,说明EC溶液微胶囊化胚芽微粉会使样品颗粒粒径提高,且随着EC溶液的升高,样品颗粒粒径提高幅度增大,符合样品密度和流动性中得出的结论,即EC溶液浓度升高,样品颗粒粒径提高。
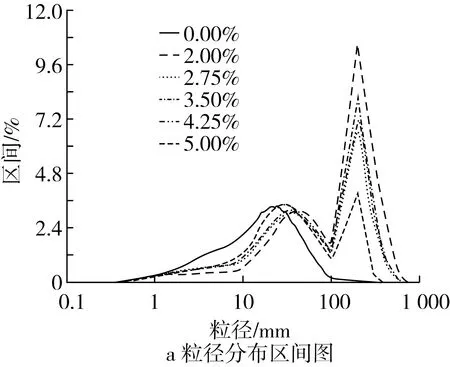

图1 胚芽系列样品粒径分布区间和累积图
中位径(D50)为样品中颗粒度小于该粒径的样品含量占样品总量的50%。D98为样品中颗粒度小于该粒径的样品含量占样品总量的98%。样品平均粒径一般考量体积、面积和长度平均径3个指标,其中体积平均径为粒径对样品体积的加权平均(三维);面积平均径,为粒径对样品表面面积的加权平均(二维);长度平均径为粒径对样品长度的加权平均(一维);比表面积单位质量样品表面积总和。由表3可知,随着EC溶液浓度的升高,样品D50和D98提高,符合样品密度和流动性中得出的结论,即EC溶液浓度升高,样品颗粒粒径提高。就样品平均粒径和比表面积而言,根据结果推断,随着EC溶液浓度的升高,样品平均粒径呈上升趋势,比表面积呈下降趋势,但根据表3中数据可知,虽样品图谱总体走向符合这一推论,但个别数据存在偏差,推测是由于个别样品均匀度较差导致的。

表3 胚芽系列样品粒径分布数据
2.3 感官评价
由表4可知,0.00%、2.00%和2.75%EC溶液微胶囊化样品评价为合格,3.50%、4.25%和5.00%EC溶液微胶囊化样品评价为良好。就样品色泽而言,样品总体呈明黄色,有较多白色掺杂其中,其中白色部分除部分膨化胚芽粉自身色泽外,EC成膜后也为白色,根据结果推断,随着EC溶液浓度升高,样品色泽分值应随之下降,但根据表4中数据,不符合这一推断,说明EC膜对样品色泽存在影响,但差别还未能被人眼察觉。就样品组织结构而言,随着EC溶液浓度升高,根据样品密度和流动性中得出的结论,即EC溶液浓度升高,样品颗粒粒径提高,均匀度下降,根据结果推断,随着EC溶液浓度升高,样品组织结构分值应随之下降,根据表4中数据,符合这一推断。就样品质地而言,由于EC溶液自身黏性的影响,样品中多为黏合物,根据结果推断,2.00%~5.00%EC溶液微胶囊化样品质地的分值差别不大,但与0%EC溶液微胶囊化样品分值差别较大,根据表4中数据,符合这一推断,2.00%~5.00%EC溶液微胶囊化样品质地的分值相近,与0.00%EC溶液微胶囊化样品存在分值差。就样品气味而言,胚芽的不良气味一直是影响其食用价值的重要因素之一,即使经过挤压膨化,其气味仍然较大,沈才洪[32]在药物微胶囊化释放特性的研究制备,采用有机溶剂蒸发技术,搅拌速度1 500 r/min,时间10 min,EC浓度为4%,明胶保护液浓度为1.5%,蒸发温度为30 ℃,制得的样品,具有明显的缓释效果,根据结果推断,随着EC溶液浓度升高,样品气味分值应随之上升,根据表4中数据,除5.00%EC溶液微胶囊化样品,其余样品均符合这一推断,其中4.25%和5.00%EC溶液微胶囊化样品气味分值相近,说明当EC溶液浓度达到4.25%以上时,对膨化胚芽粉气味可达到掩蔽效果。综合感官评价各项分值,4.25%EC溶液微胶囊化样品为最优。
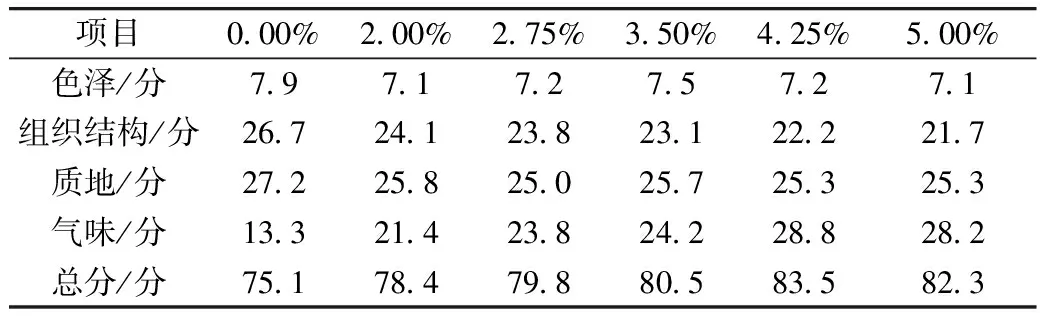
表4 胚芽系列样品感官评价
2.4 样品表面形态结构
由图2a可见,样品颗粒均匀度较差,颗粒形状多为不规则的片状聚合物,存在细长状颗粒,不同颗粒相互之间存在粘连,符合样品密度和流动性中得出的结论,即EC溶液浓度升高,样品颗粒粒径提高,均匀度下降,黏合物比例提高。

注:每组图从左到右每行依次为0.00%、2.00%、2.75%、3.50%、4.25%、5.00%EC溶液微胶囊化样品。图2 胚芽系列样品扫描电镜图
由图2b可以看出,膨化胚芽粉表面凹凸不平,整体形状不规则且有较多空洞,说明其本身就是膨化胚芽超微粉粘连成的聚合物,且结构较为松散。2.00%EC溶液微胶囊化样品表面凹凸不平,无明显空洞;2.75%EC溶液微胶囊化样品较为光滑,但存在较多褶皱;3.50%EC溶液微胶囊化样品表面较为光滑,表面凸起物较少;4.25%和5.00%EC溶液微胶囊化样品表面较为光滑,表面凸起物较多。结果表明,随着EC溶液浓度升高,芯材表面空洞被遮掩覆盖,微胶囊化样品表面逐渐光滑,但EC溶液浓度过高时,样品表面会形成大小不一的凸起物。
由图2c可见,膨化胚芽粉表面凹凸不平,存在明显的空洞;2.00%EC溶液微胶囊化样品表面出现裂纹和破损,说明出现EC膜缺损的情况,样品并未完全被EC膜包裹;2.75%EC溶液微胶囊化样品表面出现裂纹,说明样品被EC膜包裹出现,但并未达到完全覆盖的效果;3.50%EC溶液微胶囊化样品表面较为光滑,微胶囊化效果较为理想;4.25%EC溶液微胶囊化样品表面出现裂纹和破损,说明微胶囊化过程中,芯材被壁材多次包裹,料液不足导致该情况发生;5.00%EC溶液微胶囊化样品表面较为光滑,存在凸起物,说明微胶囊化过程中,芯材被壁材多次包裹。综合样品扫描电镜图分析,当EC溶液浓度达到3.50%以上时,微胶囊样品成型效果好,且不会出现EC膜未完全包裹膨化胚芽粉的情况。
2.5 加速储藏条件下样品脂肪酸值变化
由图3可知,在加速储藏条件下样品脂肪酸值都随储藏时间的延长而增加。在加速储藏条件下,其中0.00%、2.00%、5.00%EC溶液微胶囊化样品初始脂肪酸值分别为517.678、521.298、519.201 mg KOH/100 g,储藏至第30天时脂肪酸值对应增加了86.453、72.197、53.811 mg KOH/100 g。随着EC溶液浓度的升高,样品30 d内脂肪酸值增加量降低,即EC溶液对膨化胚芽粉的保护效果越明显。郑理等[31]通过采用微胶囊技术保护VC,有效提高了VC的抗氧化能力,延长其保质期。使用EC溶液微胶囊化膨化胚芽粉,可以有效减缓样品脂肪酸值增长速率。随着EC溶液浓度的升高,减缓样品脂肪酸值增长速率效果越明显。其中4.25%和5.00%EC溶液微胶囊化样品同2.00%、2.75%和3.50%样品相比,减缓幅度较为明显,且效果相近,说明当EC溶液质量浓度达到4.25%以上时,对膨化胚芽粉可达到保护效果。
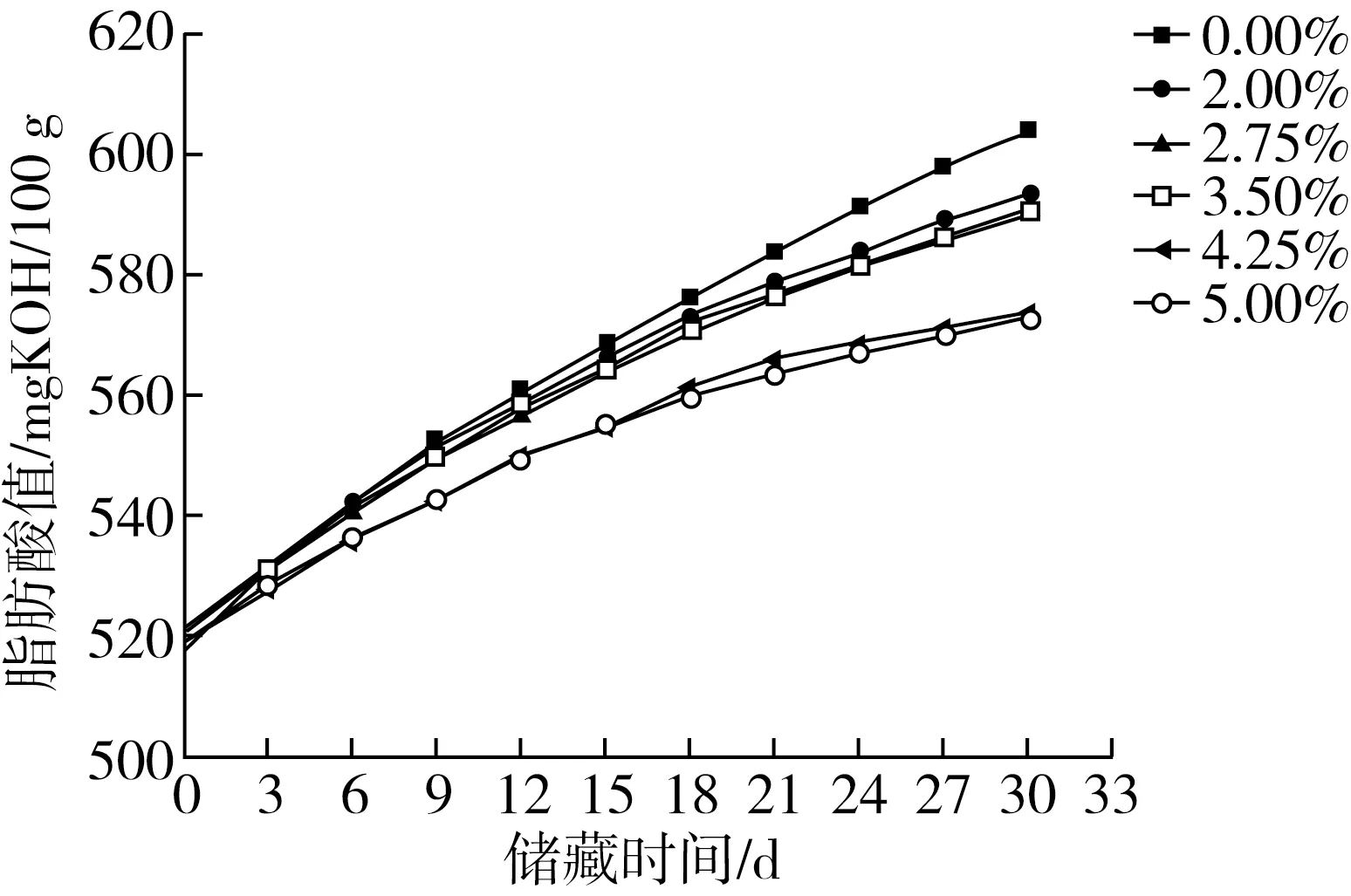
图3 加速储藏条件下胚芽系列样品脂肪酸值变化图
3 结论
选用不同浓度EC溶液为壁材,采用空气悬浮技术,对膨化小麦胚芽粉进行包埋,制得对应的微胶囊样品。为部分可溶的膨化小麦胚芽粉微胶囊化提供了参考方向。通过各微胶囊样品之间的指标比较,4.25%为EC溶液微胶囊化膨化小麦胚芽粉的最佳质量浓度。4.25%EC溶液微胶囊化样品的含水量为5.59%,溶解度为30.87%,休止角为29.71,容积密度为51.61 g/100 mL,包含率为98.14%,粒径分布均匀,结构完整,表面无裂纹或破损现象,无不良气味。此外,在加速储藏条件下,30 d内脂肪酸值增加量较小。考虑到高浓度EC溶液制得的微胶囊样品均匀度较低,后期仍需对微胶囊制备工艺进一步优化。
猜你喜欢
中国饲料(2021年17期)2021-11-02
北京建筑大学学报(2021年3期)2021-10-14
食品安全导刊(2021年21期)2021-08-30
青年文学家(2021年4期)2021-03-18
农民致富之友(2020年19期)2020-07-18
家庭百事通·健康一点通(2019年11期)2019-09-26
湖北畜牧兽医(2017年4期)2017-05-18
绿色科技(2017年7期)2017-05-12
绿色科技(2016年7期)2016-05-14
纺织导报(2014年8期)2014-10-31
