
(Fe+Co)共掺杂MgH2储氢材料的热力学稳定性和键合机理研究
2022-10-26 04:46:28黄鹏儒孙立贤
桂林电子科技大学学报 2022年3期
黄 强, 黄鹏儒, 徐 芬, 孙立贤
(桂林电子科技大学 材料科学与工程学院,广西 桂林 541004)
氢能具有绿色环保、可再生等特点,被认为是理想的二次能源。氢能的存储和运输是实现氢能应用的关键技术。氢化镁(MgH2)储氢质量分数高达7.6%,而且价格低廉,被认为是最有希望实现大规模储氢应用的材料[1-3]。
但是,氢化镁具有较高的生成焓(约-74 kJ·mol-1H2),导致其热力学稳定性较高;同时,氢化镁还具有较高的放氢反应活化能(约160 kJ·mol-1),导致其放氢反应动力学缓慢,这些缺点极大地限制了氢化镁的实际应用[4-9]。研究发现,合金化、纳米化、催化以及形成复合组织等方法能有效改善镁基储氢材料的热力学和动力学性能,氢化物的吸放氢特性在很大程度上取决于其成分,并且金属—氢键在氢化物的稳定性中起主要作用[10-13]。目前,许多研究试图通过将MgH2与过渡金属元素合金化来降低MgH2的稳定性。相关研究表明,使用非平衡处理方法将金属元素Al、Ti、Fe、Co、Ni、Cu与Mg合金化,可以有效改善MgH2的脱氢性能[10-11]。实验发现,当将FeNi纳米粒子与MgH2机械合金化时,MgH2中的H2解析温度降低至100 ℃[12],而FeCo纳米片能使MgH2中的H2解析温度降低至200 ℃,并且在室温下实现吸氢[13]。氢化物系统可以利用多组分合金共添加剂的协同效应,实现储氢性能的最优化。
拟运用第一性原理计算、研究金属Fe、Co与MgH2储氢材料共掺杂的热力学稳定性、键合机理。将MgH2的晶格原胞扩胞成3×3×1超胞,使用Fe和Co金属原子取代Mg原子,分别得到Mg12Fe2Co4H36和Mg12Fe4Co2H36双金属共掺杂体系。针对热力学稳定性问题,对共掺杂体系的晶格结构和反应生成焓进行计算、分析;结合Bader电荷和电子结构的计算,对共掺杂体系的键合机理进行探究,系统地研究共掺杂体系的储氢性能。
1 计算方法
运用基于密度泛函理论(density functional theory,简称DFT)的第一性原理计算软件VASP对MgH2及其掺杂体系进行计算[14-16]。DFT变换采用(perdew、burke、ernzerhof,简称PBE)的广义梯度近似(general gradient approximate,简称GGA)描述电子体系之间的交换关联作用[17]。采用投影缀加平面波(projector augmented wave,简称PAW)方法描述离子键与价电子的相互作用。经过收敛测试,将平面波基组扩展波函数的截断动能Ecutoff设置为500 eV,高斯展宽采用0.2 eV。晶格和原子弛豫过程中,MgH2原胞、2×2×1超胞和3×3×1超胞分别采用7×7×11、5×5×11和3×3×11的K网格点进行优化。对于MgH2原胞、超胞及其掺杂体系,统一使用自洽循环求解,Kohn-Sham方程每个原子的能量收敛值设为1×10-4eV,最大应力设置小于0.01 eV/Å。在计算中,对晶格结构充分优化,使之达到能量最小的稳定结构。对于金属原子,采用基态下稳定存在的原胞进行计算;对于H原子,选择稳定存在的氢气分子结构进行计算,使之达到稳定状态,再获取单个原子的能量。
储氢材料Mg18H36、Mg12Fe2Co4H36和Mg12Fe4-Co2H36相应的生成焓可分别表示为
ΔH(Mg18H36)=E(Mg18H36)-18E(Mg)-36E(H),
(1)
ΔH(Mg12Fe2Co4H36)=E(Mg12Fe2Co4H36)-12E(Mg)-
2E(Fe)-4E(Co)-36E(H),
(2)
ΔH(Mg12Fe4Co2H36)=E(Mg12Fe4Co2H36)-12E(Mg)-
4E(Fe)-2E(Co)-36E(H),
(3)
其中:E(Mg18H36)、E(Mg12Fe2Co4H36)、E(Mg12Fe4Co2H36)分别为Mg18H36、Mg12Fe2Co4H36、Mg12Fe4Co2H36采用离子弛豫法计算得到的超胞结构总能量值;E(Mg)、E(Fe)、E(Co)、E(H)分别为Mg、Fe、Co、H单质晶体结构中对应的单个原子的能量。
2 计算结果与讨论
2.1 MgH2及其掺杂结构的晶格结构分析
MgH2具有3种类型的晶格结构,分别是α结构、β结构和γ结构[18]。结构通常取决于温度和压力,在室温和标准气压下,MgH2常以四方对称的α结构型结晶存在,对应的空间群为P42/mnm,空间组号为136,该结构又被称为金红石型结构。在MgH2的晶胞中,2个Mg原子占据Wyckoff位置2a(0,0,0),4个H原子占据4f(0.303,0.303,0),实验测量的晶格参数分别为a=4.501 Å、c=3.010 Å[19]。α构形的MgH2晶格结构,如图1(a)所示。将金红石型MgH2晶胞沿着x轴和y轴分别扩胞成3×3×1的Mg18H36超胞,如图1(b)、(c)所示。在3×3×1的Mg18H36超胞中,有4种Mg原子和6种H原子占据不同的wyckoff位置,图1(c)分别标出了4种Mg原子和6种H原子占据的w-yckoff位置,Mg原子和H原子的坐标信息,如表1所示。为探讨FeCo双金属共掺杂对MgH2储氢材料储氢性能的影响,将Fe、Co金属原子分别任意取代金红石型Mg18H36超胞的wyckoff 2a位置中的2个Mg原子或wyckoff 4f位置中的4个Mg原子,得到Mg12Fe2Co4H36和Mg12Fe4Co2H36两种晶格结构模型,如图1(d)、(e)所示。
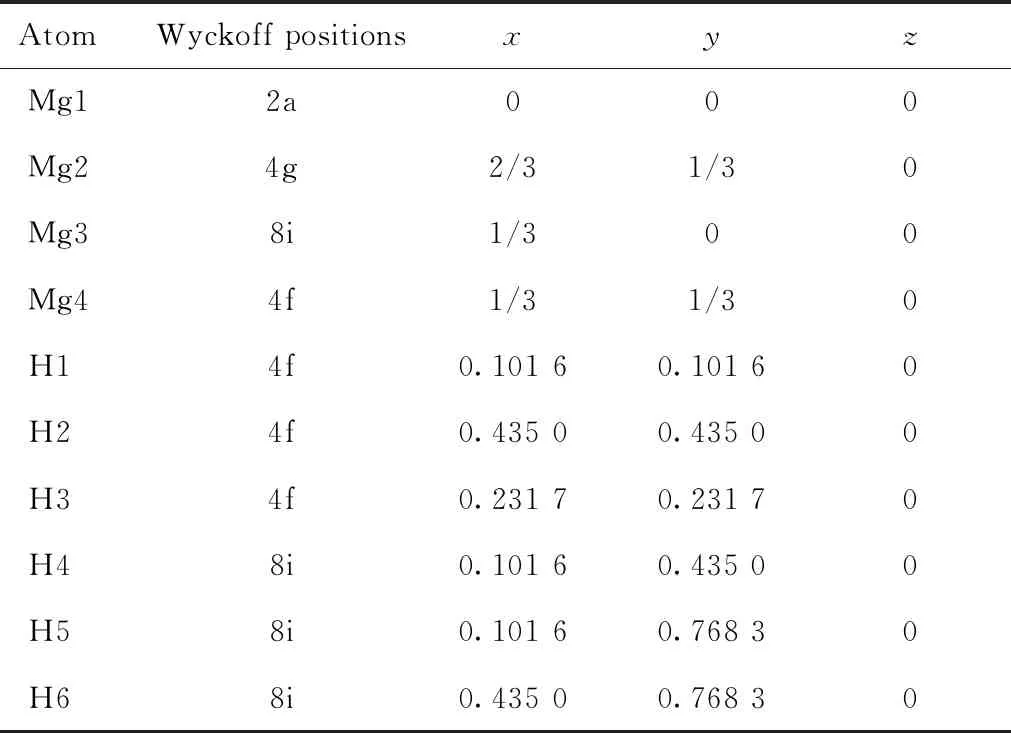
表1 Mg18H36超胞结构的初始非等价Wyckoff位置
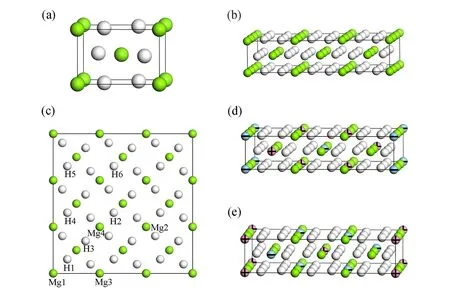
图1 晶体结构模型
图1中,(a)为MgH2晶格结构的侧视图,(b)和(c)分别为3×3×1 MgH2超胞晶格结构的俯视图和侧视图,(d)和(e)分别为Mg12Fe2Co4H36和Mg12Fe4Co2H36晶格结构的俯视图。在计算Mg18H36和(Fe+Co)共掺杂MgH2体系的总能量之前,先对所有体系的晶格结构进行结构优化,再对体系的电子结构进一步地计算。通过对比掺杂前后的晶胞结构的体积变化,发现掺杂后Mg12Fe2Co4H36和Mg12Fe4Co2H36晶胞的体积分别收缩了16%和17.5%。2种掺杂方式均呈现出收缩的趋势,晶胞体积的收缩会对晶格结构的稳定性产生影响。
2.2 (Fe+Co)共掺杂MgH2储氢材料的热力学稳性
计算得到Mg18H36、Mg12Fe2Co4H36、Mg12Fe4-Co2H36的生成焓,如表2所示。
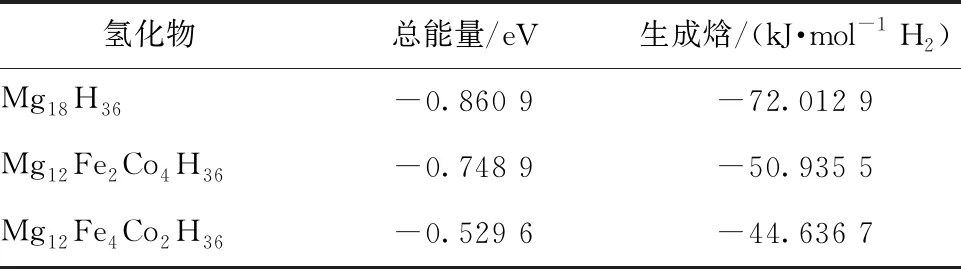
表2 Mg12Fe2Co4H36和Mg12Fe4Co2H36储氢材料氢化物的总能量和生成焓
Mg18H36及其双金属共掺杂体系的生成焓,按数值大小依次为Mg18H36>Mg12Fe2Co4H36>M-g12Fe4Co2H36。MgH2储氢材料掺杂前后的热力学稳定性,可通过金属共掺杂体系的生成焓与纯MgH2
储氢材料的生成焓之差进行评估。从能量的角度考虑,生成焓较小的体系稳定性更低,掺杂效果更好。Mg12Fe2Co4H36和Mg12Fe4Co2H36双金属共掺杂体系的生成焓数值比Mg18H36小,说明Mg12Fe2Co4H36和Mg12Fe4Co2H36双金属共掺杂体系比Mg18H36储氢材料更利于氢分子的吸附和脱附,金属Fe、Co共掺杂体系在降低MgH2储氢材料的稳定性方面起至关重要的作用,改善了储氢特性。在这2种共掺杂体系中,Mg12Fe4Co2H36的生成焓数值比Mg12Fe2Co4H36小,说明Mg12Fe4-Co2H36在降低热力学稳定性方面效果更好,更有利于提高MgH2储氢材料的储氢特性。对比Mg18H36及其掺杂体系的生成焓值,表明(Fe+Co)双金属共掺杂是降低MgH2储氢材料热力学稳定性的有效方法。
2.3 (Fe+Co)共掺杂MgH2储氢材料的键合作用
2个成键原子的键长可直接用于评估原子间的粘合强度。在Mg18H36及其掺杂体系中,金属原子间的键长如表3所示。
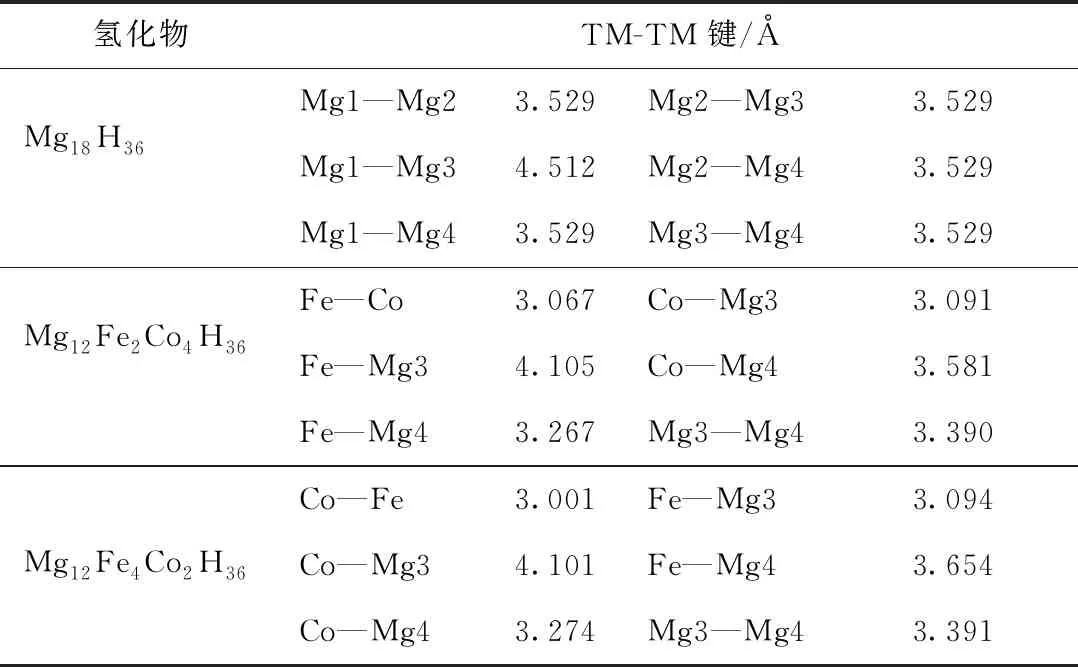
表3 Mg18H36及其掺杂体系的金属与金属之间的键长
在Mg18H36结构中,Mg1—Mg3键长为4.51 Å,其他Mg原子间的键长为3.53 Å,键长均值为3.70 Å,这与其他理论结果吻合得很好[19-20]。在金属共掺杂体系中,金属原子间所有键长都比纯Mg18H36体系的短,处于3.00 ~ 4.11 Å之间。Mg12Fe2Co4H36、Mg12Fe4Co2H36掺杂体系的金属间键长的均值依次为3.417 Å、3.419 Å,其中Fe—Co、Co—Fe键的键长依次为3.067 Å、3.001 Å,与Mg18H36结构中的Mg1—Mg2键相比,分别缩短了13.09%、14.96%。此外,通过对晶格结构进行分析发现,除了Mg12Fe2Co4H36结构中的Co—Mg4和Mg12Fe4Co2H36结构中的Fe—Mg4键延长之外,其他金属键都缩短了。就整体而言,金属共掺杂后金属键呈现出缩短的趋势,意味着金属原子间的键合作用显著增强,掺杂体系的合金化趋势得以提高。因此,进一步可以推断出,这些增强的金属原子之间的键合作用破坏了原有体系的平衡,致使氢原子和金属原子之间的键合作用被削弱。
为分析Mg18H36储氢材料及其掺杂体系中H原子和Mg原子之间的键合作用,将Mg18H36超胞中的原子以Mg原子为中心,划分为4种八面体结构,如图2所示,(a)、(b)、(c)和(d)分别为Mg18H36结构中Mg1、Mg2、Mg3和Mg4原子与近邻的6个H原子组成的八面体结构。
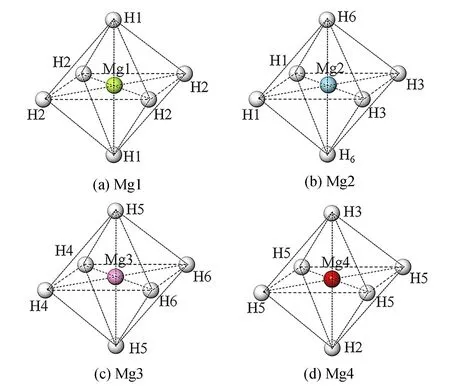
图2 4种不同八面体结构
在Mg18H36超胞中,金属原子及其近邻的6个H原子可看作是1个八面体构型,不同金属原子近邻的H原子则不同。
对于Mg12Fe2Co4H36和Mg12Fe4Co2H36掺杂结构,Fe、Co原子依次取代Mg1和Mg2原子的位置,且以共价键的方式与H原子结合,形成TM—H键,TM—H键的键长,如表4所示。
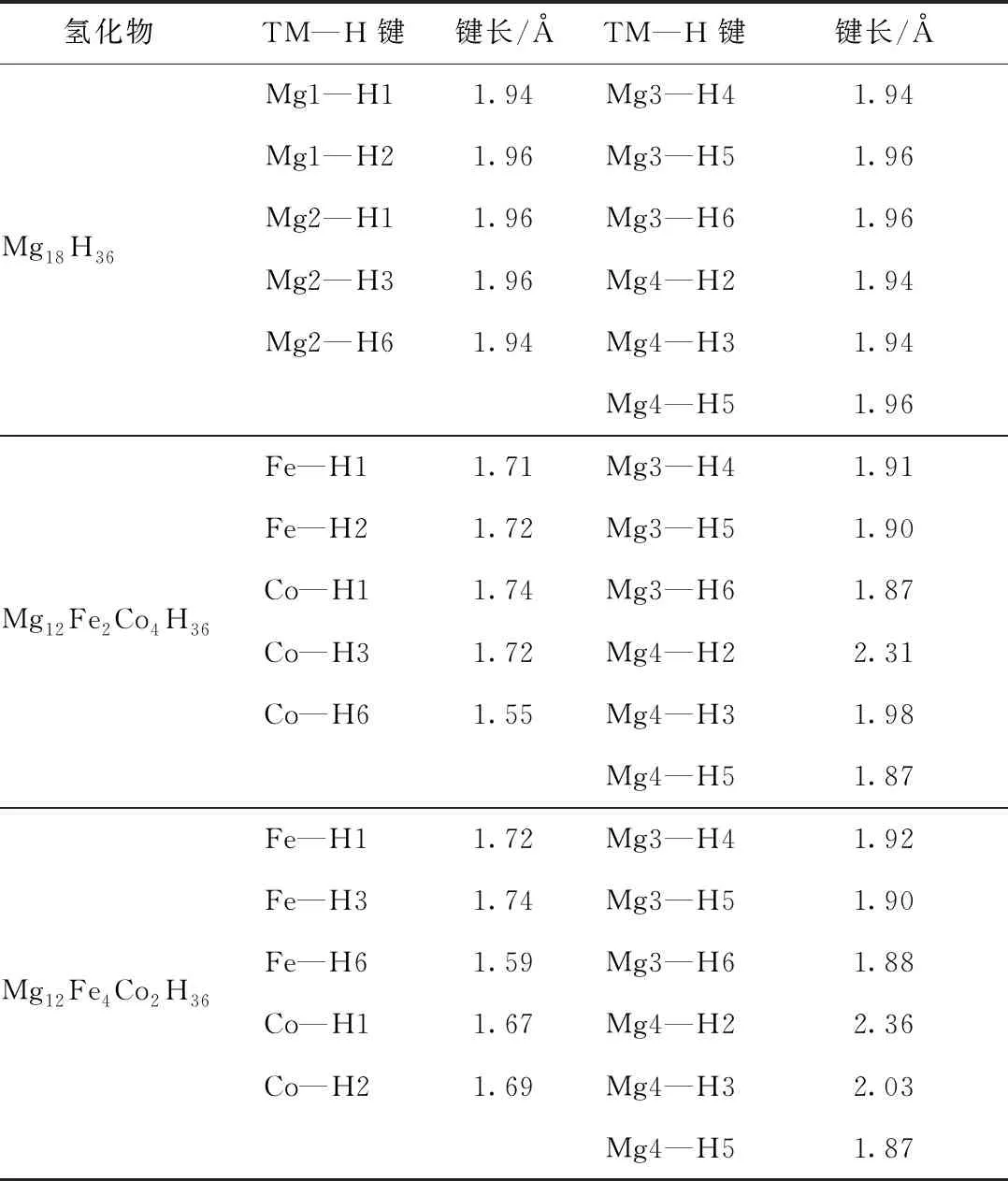
表4 Mg18H36、Mg12Fe2Co4H36和Mg12Fe4Co2H36晶格结构中金属原子和近邻氢原子的键长
计算结果表明,Mg18H36结构的键长范围为1.940~1.959 Å,Mg12Fe2Co4H36和Mg12Fe4Co2H36结构的键长范围分别为1.550~2.134 Å、1.585~2.357 Å。与纯的MgH2体系相比,Mg12Fe2Co4H36和Mg12Fe4Co2H36共掺杂体系中Fe—H键和Co—H键键长缩短了10%以上,而Mg3—H键缩短了1.03%~4.59%。TM—H键大体上呈现出键长缩短的趋势,然而Mg4—H2键和Mg4—H3键却呈现出增长的趋势。因此,就整体而言,金属共掺杂降低了超胞的体积,但Mg—H键的键长并未因此而显著减少,相反部分Mg—H键的键长却增加了。结果表明,H原子更趋向与掺杂的金属原子结合成键,削弱H原子与Mg原子的键合作用,使Mg—H键增长,键的强度变弱,从而进一步降低氢的解吸温度。
为研究Mg18H36储氢材料及其掺杂体系中氢原子和金属原子之间的键合作用,对Mg18H36储氢材料及其掺杂体系进行Bader电荷分析。Bader额外电荷(简称BEC)指Bader原子中所含电荷与原子电荷之差。通过比较BEC,可以在一定程度上估计化学键的离子化程度。Mg18H36及其掺杂系统的BEC值如表5所示。
BEC正值越大,原子的阳离子性就越强;BEC负值越接近零,原子的阴离子就越少。由表5可知,对于纯MgH2系统而言,Mg和H原子的BEC值表明Mg和H之间的化学键都是强离子性的。
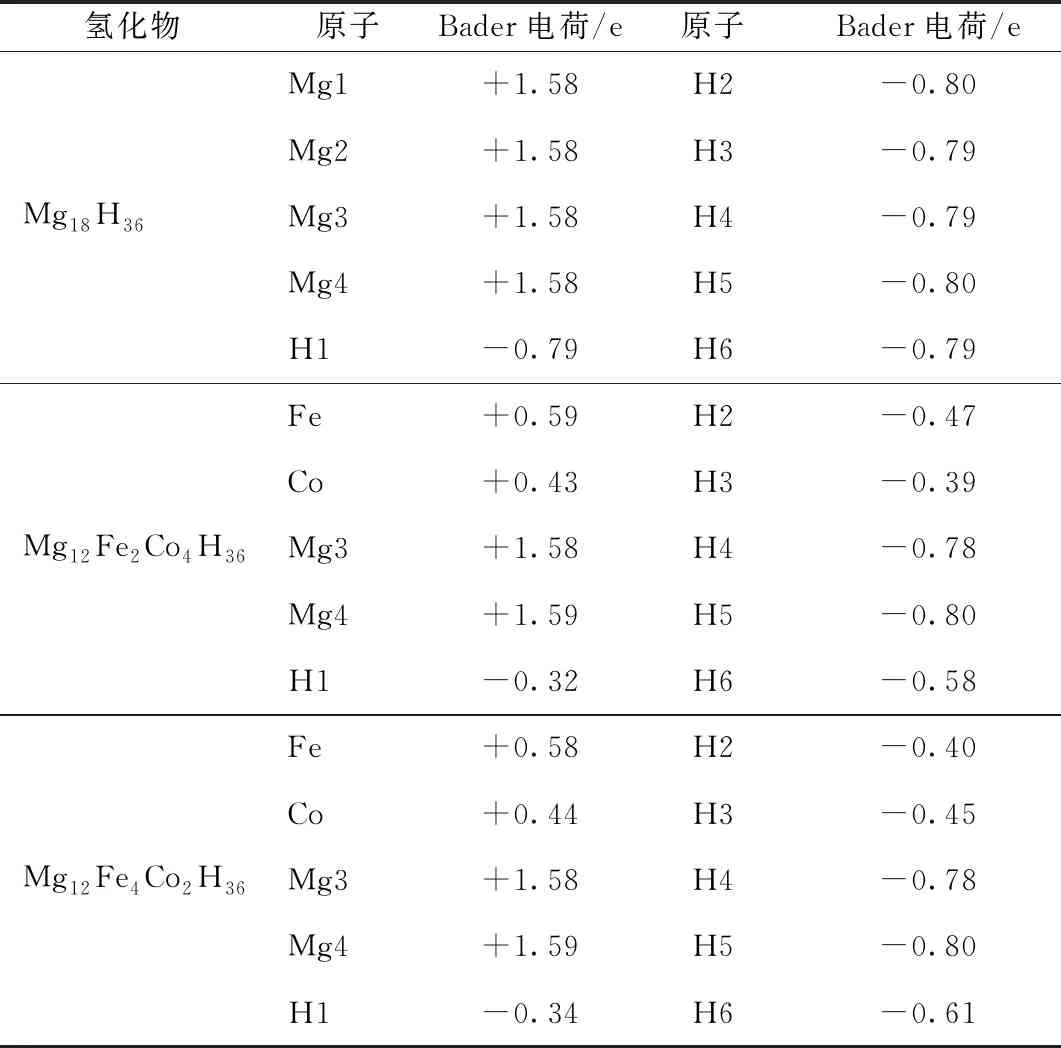
表5 Mg18H36、Mg12Fe2Co4H36和Mg12Fe4Co2H36晶格结构中各原子的Bader电荷值
当Fe和CO共掺杂时,除H4、H5之外H原子的BEC绝对值都大大降低,Mg原子的BEC值比纯体系的BEC值略有增加,且比Fe和Co原子的BEC值大。此外,H1,H2,H3和H6原子的BEC的绝对值较小,意味着Fe—H键和Co—H键的离子性较弱,因此掺杂体系中的离子键强度也是减弱的。掺杂体系中H原子的BEC的绝对值较小,在于与Mg原子相比,Fe、Co和Ni原子的BEC值较少,而原子的BEC值与其电负性密切相关。与纯MgH2相比,掺杂体系中Mg3和Mg4原子的BEC值略大,且H4和H5原子的BEC值数值上也较大,表明Mg3—H4,Mg3—H5和Mg4—H5的离子键强度略有增强。
2.4 (Fe+Co)共掺杂MgH2储氢材料电子结构分析
为理解氢化镁的热稳定机理,对Mg18H36及其共掺杂体系的电子结构进行深入研究。纯MgH2的电子态密度(简称DOS)如图3所示。
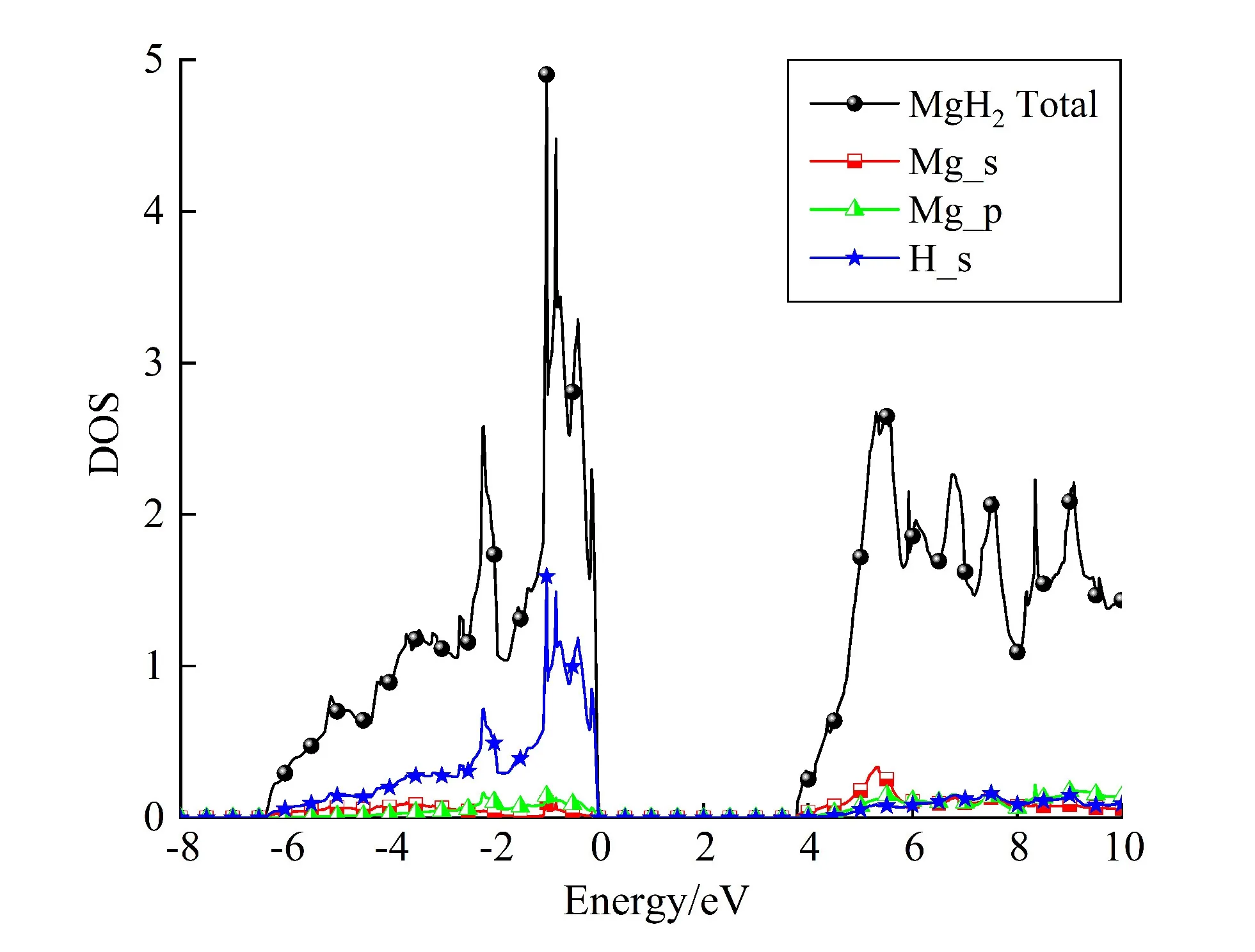
图3 MgH2储氢材料的电子态密度
MgH2的价带与导带之间存在着较大的带隙,是一个绝缘体,计算得到的带隙值为3.74 eV。价带主要由H_s轨道贡献,而导带主要由Mg_s和Mg_p轨道贡献,意味着在Mg原子与H原子之间,由于电荷从Mg原子转移到H原子,形成强的离子键。此外,通过分析Mg_s和Mg_p轨道可知在价带中Mg_s和Mg_p轨道与H_s轨道价键的相互作用。因此,研究人员通常认为,在MgH2储氢材料中,Mg—H键的键合机理为离子键和共价键的共同作用。另一方面,在MgH2中存在相对较大的带隙,容易使Mg—H键发生解离。为了在脱氢过程中恢复Mg的金属特性,需将电子从H原子逆向转移到Mg原子。因此,为了释放氢,纯氢化镁必须获取更多能量,从而为电子迁移提供充足的能量。这也正是纯氢化镁具有热力学稳定性且分解温度非常高的原因。
(Fe+Co)双金属共掺杂系统总的DOS和各原子的DOS如图4所示。总的DOS和金属原子的DOS绘制在左侧,H原子的DOS(H1—H6)绘制在右侧。观察掺杂体系的DOS发现,掺杂体系的带隙皆消失,带隙值都为0 eV,这表明掺杂之后的材料展现出了金属特性。价带及靠近费米能级附近的价带主要由掺杂金属原子的d轨道贡献,尤其是费米能级附近的价带一侧,贡献峰的峰值均较大。金属原子d轨道的贡献是使掺杂体系的带隙消失,将氢化镁储氢材料转变为导体。
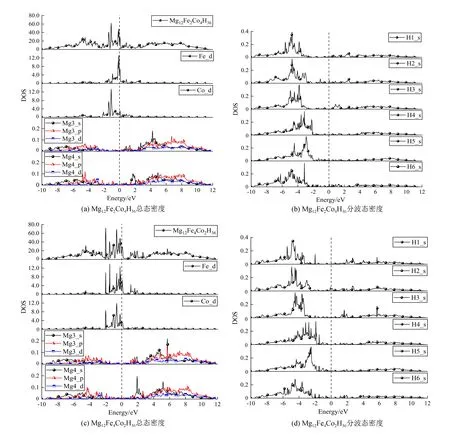
图4 Mg12Fe2Co4H36、Mg12Fe4Co2H36的总态密度和分波态密度
由于脱氢过程涉及加热,吸放氢温度是衡量氢化物储氢性能的重要指标。Fe和Co金属的共掺杂,使MgH2储氢材料从绝缘体转变成具有金属性质的材料,进而提高MgH2储氢材料的导热性能,这将有利于氢气的解析。除了在费米能级附近存在较强的峰以外,在-6.1 ~ -2.5 eV的能量区域还集中分布着一定强度的峰。H1、H2、H3、H6原子的最高电子态密度峰值位于-4.9 ~ -3.5 eV,恰好与Fe和Co原子的d轨道交叠。因此,轨道杂化主要发生在Fe、Co原子的d轨道与H1、H2、H3、H6原子的s轨道之间。轨道的杂化使得原子间成键,如表4所示。掺杂金属与H原子之间存在较短的键长,便是轨道杂化的结果。
Mg原子的p轨道存在较宽的能量范围,H4和H5的能量介于-3 ~ -2.7 eV之间,与Mg_p和H_s轨道存在良好重叠。因此,H4和H5原子的s轨道主要与Mg_p轨道杂化有关。在掺杂系统中,电子很容易从H原子转移到Mg原子,同样,金属原子与H原子之间的键也容易发生断裂。对比掺杂前后的电子态密度可知,图4所示的H和Mg原子的电子分波态密度在价带区域,整体上小于图3所示的H和Mg原子的分波态密度,表明Mg与H之间的键相互作用较弱。在金属共掺杂体系中,Mg原子与H原子之间的键合作用减弱。就杂化效应而言,在共掺杂体系中,Mg原子与H原子的轨道杂化比纯MgH2体系的弱,这说明Mg—H键结合强度减弱,与表5中掺杂体系的Mg—H键键长较长相一致。
因此,通过分析Mg18H36及其共掺杂体系的电子结构可知,(Fe+Co)双金属共掺杂对MgH2储氢材料热力学性能的改善有积极影响。
3 结束语
运用第一性原理研究了(Fe+Co)双金属共掺杂MgH2储氢材料,扩展到三元氢化镁,阐明多组分系统中合金元素与氢原子之间的键合性质以及掺杂系统的电子结构。在共掺杂系统中,发现Fe、Co的掺杂可降低MgH2储氢材料的生成焓,从而达到有效降低MgH2储氢热力学稳定性的效果。通过对MgH2掺杂前后的键长以及Bader电荷的分析,发现金属的掺杂整体上起到了弱化Mg—H键的作用。电子结构的计算分析表明,掺杂后Mg—H键结合强度减弱,(Fe+Co)双金属共掺杂对MgH2储氢材料热力学性能的改善有积极影响,可为发展高性能储氢材料提供理论指导。
猜你喜欢
中国特种设备安全(2022年4期)2022-07-08 02:41:40
中国特种设备安全(2022年4期)2022-07-08 02:41:28
航天工业管理(2020年9期)2020-12-28 00:38:34
重型机械(2020年2期)2020-07-24 08:16:08
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
石油化工建设(2018年2期)2018-07-11 01:25:06
信息记录材料(2016年4期)2016-03-11 15:22:31
材料科学与工程学报(2016年5期)2016-02-27 07:11:37
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
