
炎症在肝纤维化中的作用
2022-10-19 02:44刘华宝胡文艳饶春燕
临床肝胆病杂志 2022年10期
李 婷, 刘华宝, 胡文艳, 饶春燕
重庆市中医院 肝病科, 重庆 400021
肝纤维化是由多种因素引起的慢性肝损伤所导致的细胞外基质(extracellular matrix,ECM)不断积累,形成瘢痕组织的病理过程。主要表现为肝星状细胞(hepatic stellate cell,HSC)被激活转化为肌成纤维细胞(myofibroblasts,MF),ECM过度沉积,替代受损的肝组织[1]。慢性肝损伤引起的细胞死亡和炎症是肝纤维化发展的2个核心因素[2]。了解炎症信号在肝纤维化中的作用,有利于肝纤维化的临床诊断、治疗以及新药的研发。本文将从炎症与肝纤维化间的联系机制着眼,重点介绍肝脏炎症信号的产生,以及HSC、Kupffer细胞、炎症小体等在肝纤维化过程中的作用。
1 肝纤维化中炎症信号的产生
肝纤维化多由肝毒性损伤和胆汁淤积性损伤这两种常见的慢性肝损伤引起。肝毒性损伤是由HBV、HCV感染以及代谢综合征诱发的非酒精性脂肪性肝炎(NASH)导致的。胆汁淤积性损伤是由原发性胆汁性胆管炎、原发性硬化性胆管炎、胆道闭锁等疾病导致的胆汁流动受阻引起的[1]。病毒、细菌、胆汁酸、酒精代谢物等肝毒性物质引发肝细胞损伤和死亡[3]。损伤的肝细胞中核因子-κB(NF-κB)等信号通路被激活,诱导TNFα、IL-6和CC类趋化因子配体2(chemokine CC motif ligand 2,CCL2)等促炎细胞因子和趋化因子的释放,诱发炎症反应[4]。同时,细胞坏死、坏死性凋亡以及细胞焦亡等形式的肝细胞死亡也会伴随大量炎症介质的释放,加剧炎症反应,进一步激活HSC和Kupffer细胞引发肝纤维化(图1)[5]。研究[6]表明,NASH患者肝细胞的死亡程度与肝纤维化的严重程度密切相关。
此外,损伤或死亡的肝细胞释放损伤相关模式分子(damage-associated molecular pattern,DAMPS)也在纤维化的发展和炎症中发挥重要作用。DAMPS不仅直接激活HSC(图1),还可诱发无菌炎症进一步导致肝细胞损伤[7]。DAMPS是一种内源性分子,主要包括高迁移率族盒蛋白1(high mobility group box-1,HMGB-1)、双链/单链的DNA或RNA、线粒体DNA、三磷酸腺苷(adenosine triphosphate,ATP)等[8]。HMGB-1是一种非组蛋白,在真核细胞中广泛表达。坏死的肝细胞分泌的HMGB-1能够与Toll样受体(Toll-like receptors,TLR)4、9结合,激活NF-κB信号而促进炎症反应,还可与单链NDA、脂多糖(lipopolysaccharide,LPS)、IL-1β等其他炎症因子形成复合物,启动炎症反应[9-10]。而且,HMGB-1对中性粒细胞在肝损伤部位的募集也有一定的影响[11]。HMGB-1还能够激活HSC,促进α平滑肌肌动蛋白(alpha-smoothmuscleactin,α-SMA)生成并抑制基质金属蛋白酶-2的表达,增加ECM的沉积,加重肝纤维化[12]。肝细胞死亡或损伤释放ATP,而细胞外高浓度的ATP刺激嘌呤能受体P2X7激活核苷酸结合寡聚化结构域样受体蛋白3(nucleotide binding oligomerization domain-like receptor protein 3,NLRP3)炎症小体,产生具有活性的IL-1β[13]。在细胞损伤的情况下,组蛋白也会释放至细胞外,起到DAMPS的作用,与TLR2/4结合,通过促进NLRP3炎症小体的形成诱导无菌炎症[14]。过氧化物还原酶-1(peroxiredoxin-1,Prdx1)是一种新发现的与肝损伤有关的DAMPS,Prdx1与TLR2/4结合激活NLRP3炎症小体介导炎症反应的发生,同时,Prdx1还通过激活NF-κB和NLRP3炎症小体,参与IL-1β、IL-6和TNFα等炎症因子的产生[15-16]。
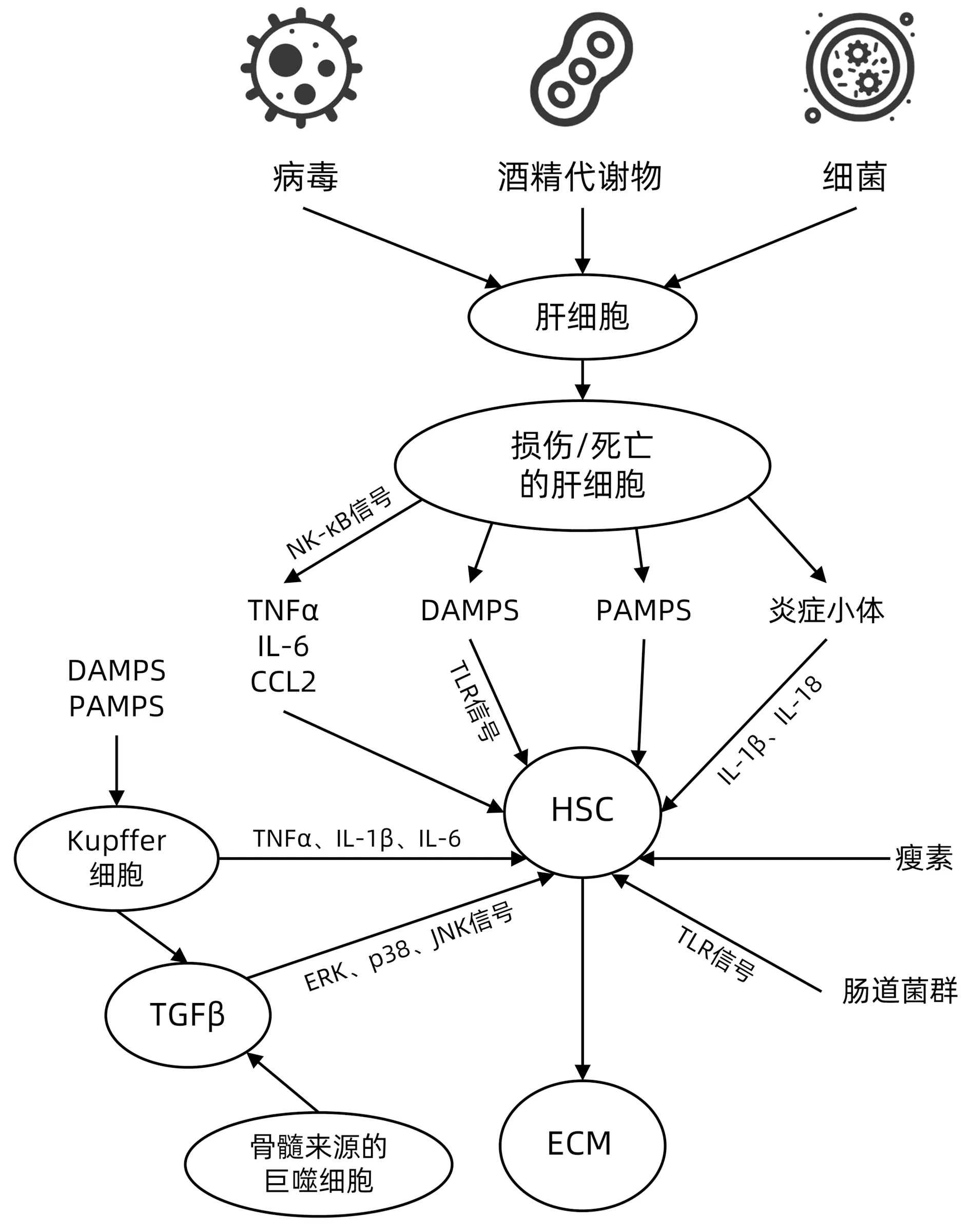
图1 炎症信号激活HSC机制图
死亡的肝细胞也可通过其他方式介导炎症反应以及HSC的激活。研究[17]表明,肝细胞凋亡所产生的凋亡小体被HSC吞噬后激活HSC(图1),并上调前胶原α1、TGFβ1、NADPH氧化酶的表达以及胞内活性氧(reactive oxygen species,ROS)水平。ROS可直接或间接激活NLRP3炎症小体,促进炎症反应的发生[18]。
2 联系炎症与肝纤维化的细胞和炎症介质
2.1 肝星状细胞(HSC) HSC是肝纤维化中炎症信号的主要接收细胞和促纤维化的主要效应细胞[19]。肝细胞损伤、死亡引发炎症反应,炎症激活HSC引起ECM的分泌和沉积而导致肝纤维化。正常生理状态下,HSC位于肝Disse腔中,处于静止状态,胞浆内含有大量的维生素A脂滴。HSC被激活后,胞内维生素A含量减少,脂肪生成表型丧失,转化为具有增殖和收缩能力的MF。在这个过程中,活化的HSC迁移到损伤部位,产生大量的ECM和炎症介质,Disse腔中的IV型胶原蛋白被纤维状的Ⅰ型和Ⅲ型胶原蛋白所取代,形成瘢痕组织。此外,激活的HSC表达高水平的α-SMA和金属蛋白酶组织抑制因子-1(tissue inhibitors of metalloproteinase-1,TIMP-1),促使其从脂肪细胞表型向促纤维化和炎症表型的转变[4,19]。在慢性损伤性肝病中,受损及死亡的肝细胞、Kupffer细胞、骨髓来源的巨噬细胞和胆管细胞等分泌的炎症介质持续刺激HSC,导致肝纤维化程度不断加重。HSC还通过其表面的TLR接收肠源性信号促进其活化和纤维化[20]。在肥胖症中,瘦素也能介导HSC的激活,瘦素基因缺陷大鼠能够抵抗CCl4诱导的肝纤维化[4]。
2.2 Kupffer细胞 Kupffer细胞是肝脏中固有的巨噬细胞,主要位于肝血窦中,占体内巨噬细胞的80%~90%,在清除和防御胃肠道来源的细菌及致病物质中发挥重要作用[21]。在肝损伤的情况下,Kupffer细胞被DAMPS或LPS、病毒DNA等病原相关模式分子(pathogen-associated molecular patterns,PAMPS)激活,促使Kupffer细胞中炎症小体的合成和活化以及IL-1β、IL-18、CCL2的释放,这些改变将导致循环中白细胞(单核细胞和中性粒细胞)的募集和肝窦内皮细胞血管细胞黏附分子的增加[19]。激活的Kupffer细胞还可与TLR、甘露糖受体和Nod样受体等模式识别受体结合刺激HSC活化[3,21]。有研究[22-23]发现,Kupffer细胞被LPS激活后与TLR4结合,产生TNFα、IL-1β、IL-6、IL-12、IL-18激活HSC(图1)。
肝纤维化中,TNFα、IL-1β通过上调TIMP-1,下调骨成形蛋白-激活素膜结合阻断因子(BMP and the activin membrane-bound inhibitor,BAMBI)并抑制ECM的分解,阻止HSC凋亡而加重纤维化程度。有研究表明,IL-1β的基因敲入小鼠会发生自发性肝损伤和肝纤维化[20],而IL-1β基因敲除小鼠则能预防脂肪性肝炎和肝纤维化的发生[24]。TNFα和肿瘤坏死因子受体1(tumor necrosis factor receptor type 1,TNFR1)基因缺陷小鼠能减轻胆汁淤积导致的肝纤维化[20]。而IL-6作为一种经典的促炎因子,常被用于临床判断慢性肝纤维化的活动程度[25]。
2.3 骨髓来源的巨噬细胞 巨噬细胞分为促炎的M1型与抗炎的M2型,M1巨噬细胞在LPS、TNFα和干扰素γ的诱导下,表达TNFα、IL-6和IL-1,参与慢性肝炎的发病过程。M2巨噬细胞在IL-4、IL-10、IL-13诱导下产生IL-10、TGFβ、血小板衍生生长因子(Platelet derived growth factor,PDGF)以及表皮生长因子发挥抗炎和促进创面愈合的作用[26-27]。Kupffer细胞和骨髓来源的巨噬细胞均高度表达TGFβ(图1),表明这2种细胞均具有促肝纤维化的作用[28]。TGFβ是肝纤维化过程中关键的调节因子。TGFβ能够激活细胞外调节蛋白激酶、p38以及c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)等3条丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路,激活HSC29]。TGFβ1还可通过激活Notch通路、增加α-SMA的表达促进HSC向MF转化[30]。在活化的HSC中,TGFβ通过Smad依赖途径诱导Ⅰ型和Ⅲ型胶原蛋白的转录,但同时也会抑制HSC的增殖[31]。此外,TGFβ还抑制了自然杀伤细胞(natural killer cell,NK)的活性,阻止NK细胞诱导的HSC凋亡,影响NK细胞的抗纤维化功能[32]。
研究[33-34]发现,在动物模型中CCL2和其他趋化因子可促进CCR2+/Ly-6Chigh单核细胞募集到肝脏中的受损部位,转化为促进炎症、血管生成和纤维生成的 Ly-6C+巨噬细胞。Ly-6C+巨噬细胞可通过释放TGFβ1和PDGF刺激 HSC向 MF转化。促纤维化的Ly-6C+巨噬细胞可通过激活NF-κB信号来提高MF的存活,还可通过CCL2实现MF的募集。
2.4 炎症小体 炎症小体是一种由Nod样受体、凋亡相关的斑点样蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)和半胱氨酸天冬氨酸蛋白酶1(cysteine-containing aspartate-specific proteases,Caspase-1)组成的蛋白复合物。模式识别受体通过识别的DAMPS或PAMPS促进炎症小体的组装和激活,活化的炎症小体可诱导IL-1β前体和IL-18前体的翻译转录及Caspase-1活化[35]。炎性小体及其下游效应因子在肝纤维化发展中发挥着不可替代的作用[2]。在实验性肝纤维化模型中,NLRP1、NLRP3和黑色素瘤缺乏因子2炎性小体在Kupffer细胞和HSC表达均显著上升[36]。小鼠NLRP3炎症小体的持续激活可导致严重的肝脏炎症、纤维化和肝细胞凝固性坏死[37]。在NASH小鼠模型中,NLRP炎症小体的激活诱导纤维化反应[38]。另有研究[39]发现,NLRP3炎症小体以及其组成部分(Caspase 1、NLRP3、ASC)在慢性HCV患者和NASH患者中均明显升高,且与TIMP-1、α-SMA、Ⅰ型胶原蛋白的表达具有相关性;肝纤维化患者的肝组织活检中也发现α-SMA阳性,细胞中NLRP3的表达升高。以上研究表明,炎症小体可能是诱导纤维化发展的关键因素。
2.5 IL家族
2.5.1 IL-17 IL-17由辅助性T淋巴细胞17(T helper cell 17,Th17)分泌,在病毒性肝炎、酒精性肝病和自身免疫性肝炎中呈高表达。在实验性肝纤维化模型中,IL-17A通过激活NF-κB和转录激活因子3(signal transducer and activator of transcription 3,STAT3)刺激Kupffer细胞和HSC产生IL-6、TNFα与TGFβ。IL-17还可通过STAT3依赖途径直接诱导HSC活化。IL-17A和IL-17RA缺陷小鼠均显示肝纤维化减轻[40-41]。
2.5.2 IL-20 IL-20是一种促纤维化的细胞因子,其含量在患有肝纤维化的人和小鼠中显著上升。IL-20可促进HSC的激活、增殖和迁移。抑制IL-20及其受体,不仅可以减轻肝纤维化,还可以减轻肝损伤,提示IL-20不仅可作用于HSC,还可作用于肝细胞[42]。
2.5.3 IL-33 IL-33是IL-1家族成员,主要诱导Th2型免疫应答。正常情况下,IL-33位于细胞核中发挥转录因子的作用。当细胞受损后IL-33被分泌至胞外,与生长刺激表达基因2蛋白(growth STimulation expressed gene 2,ST2)结合,从而作为细胞因子发挥作用。 IL-33和ST2在小鼠及人肝纤维化中的表达显著上升。慢性肝损伤诱导肝细胞分泌IL-33,进而刺激Ⅱ型固有淋巴细胞(Type Ⅱ innate lymphoid cells,ILC2)产生IL-13。IL-13通过IL-4Rα和STAT6促进HCS活化。IL-33、ILC2基因缺陷小鼠均表现出抗肝纤维化能力[43]。
3 肝纤维化中的炎症信号通路
3.1 TLR信号通路 TLR家族具有识别病原体的能力,包含10个成员:TLR1~10。其中TLR1、2、4、5、6位于细胞膜上,TLR3、7、8、9和10位于细胞内溶酶体中。TLR及其配体结合会激活肝纤维化通路[44]。TLR2及其配体在HBV诱导的肝纤维化中刺激Kupffer细胞分泌IL-10,而且TLR2与TLR1或TLR6可以以同源二聚体或异源二聚体的形式通过MyD-88激活NF-kB信号。外泌体介导的TLR3信号通路可增加IL-17的分泌,促进肝纤维化。在NASH大鼠模型中,TLR4-p38MAPK信号可诱导Kupffer细胞活化。TLR2和TLR9以MyD88依赖途径激活HSC分泌CXC趋化因子配体1。TLR7可刺激树突状细胞分泌干扰素,激活Kupffer细胞产生促纤维化的IL-1受体拮抗剂[45]。
3.2 NF-κB信号通路 NF-κB是一种核转录因子,作为炎症和细胞死亡的关键调节因子,在许多慢性肝病中发挥重要作用。NF-κB信号可被TLR、IL-1β、TNFα等多种炎症介质、信号通路和细胞应激激活,抑制NF-κB信号可有效促进细胞死亡,抑制炎症和纤维化[46]。HSC中NF-κB的激活导致HSC存活率增加,促进纤维生成。抑制Kupffer细胞中的NF-κB信号可减轻CCl4诱导的肝纤维化[20,33]。此外,NF-κB激活导致HSC分泌趋化因子,使TLR4和TNFα介导的BAMBI下调,增加HSC对TGFβ的敏感性,从而增强纤维化[47]。
3.3 JNK信号通路 JNK是MAPK家族成员,JNK信号能够被TLR、IL-1β、TNFα、ROS和饱和游离脂肪酸等激活,在肝细胞损伤、代谢、炎症和纤维化等方面发挥重要作用[48]。在HSC中,JNK信号能通过促进PDGF、TGFβ和血管紧张素Ⅱ诱导α-SMA以及胶原蛋白的生成发挥促纤维化作用,也可通过TGFβ和PDGF介导的Smad依赖途径诱导胶原蛋白的生成,加重纤维化程度[49-50]。
4 结语
肝纤维化是一种肝脏应对慢性损伤自我修复的过程。正常的炎症反应有利于肝损伤部位的愈合,但在慢性刺激下,持续的炎症刺激会导致肝纤维化发展为肝硬化或肝细胞癌等不可逆的肝损伤。肝纤维化过程受炎症信号和肝细胞、Kupffer细胞及HSC间的相互作用。肝毒性物质导致肝细胞受损或死亡,释放内源性危险信号。Kupffer细胞及其他炎症细胞会被肝毒性物质或DAMPS激活促进炎症细胞因子的释放,并进一步作用于所有其他类型的细胞,以维持炎症状态。炎症信号激活HSC并促使其向分泌ECM且高度增殖的MF转化。同时,无菌炎症和炎症小体的激活,也进一步促进纤维化形成与炎症反应的延续。虽然炎症反应是导致肝纤维化发生发展的重要因素,但目前以炎症信号为靶点的肝纤维化临床治疗药物却相对匮乏。尽管在动物实验模型中,IL-6、TNF、TGFβ抑制剂等多种药物已成功逆转肝纤维化,使纤维化组织得以消退。但上述药物存在严重的副作用,例如抑制TNF可能会增加炎症导致的风险,而长期阻断TGFβ可能会抑制伤口愈合并促进肿瘤的发生。由于这些细胞因子的功能对于维持体内免疫反应,组织修复平衡至关重要,因此,长期靶向性的抑制药物的研发仍具有挑战性。最新研究发现的几种新的抑制肝脏炎症药物,如血清淀粉样蛋白P,其作为五聚体蛋白家族的一员,由肝脏产生并分泌到血液中,可抑制单核细胞分化为纤维细胞,减少中性粒细胞对ECM蛋白的黏附,降低促炎巨噬细胞的激活,并促进细胞碎片的吞噬[1]。近年来,中医药也因为其多成分、多靶点的特点在肝纤维化的治疗中备受关注。未来关于炎症和纤维化的研究应侧重于确定“核心途径”,不仅可以提高对这一复杂网络的理解,还可以将治疗重点放在特定的候选分子上,例如关键的上游调节因子或转录主调控因子。炎症引发肝纤维化的过程错综复杂,炎症对纤维化的影响仍需要不断的探索与研究。进一步了解炎症相关细胞、细胞因子和信号通路在肝纤维化中的相互作用,有助于肝纤维化发病机制的阐明和临床新药的研发。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:李婷负责撰写论文;李婷、刘华宝、胡文艳负责查找文献,分析资料;饶春燕负责拟定写作思路,指导论文撰写。
猜你喜欢
浙江医学(2022年18期)2022-11-05
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国现代医生(2022年21期)2022-08-22
中国药学药品知识仓库(2022年9期)2022-05-23
中国药学药品知识仓库(2022年9期)2022-05-23
中国典型病例大全(2022年13期)2022-05-10
当代陕西(2022年5期)2022-04-19
家庭医学·下半月(2022年3期)2022-04-07
科学与财富(2021年33期)2021-05-10
