
卡巴拉汀合成技术综述
2022-10-18 03:54耿梅林子婷国家知识产权局专利局专利审查协作江苏中心江苏苏州215163
化工管理 2022年28期
耿梅,林子婷(国家知识产权局专利局专利审查协作江苏中心,江苏 苏州 215163)
0 引言
阿尔茨海默症 (alzheimer’s disease, AD),俗称老年痴呆,是一种多发于老年人的中枢神经系统进行性退行性疾病,其表现症状为进行性记忆、认知能力功能障碍。AD患者认知损伤程度各有不同,但对病人日常活动及家庭生活质量均会产生诸多影响。目前AD发病原因不明确,无法治愈,药物干预延缓病情发展是主要的治疗手段,研究最多并广泛用于临床的是乙酰胆碱激酶抑制剂代表药物之一即为卡巴拉汀(rivastigmine)。
卡巴拉汀,化学名N-甲基乙基氨甲酸-3-[(1S)-1-(二甲胺基)乙基]苯酯。研究表明,卡巴拉汀进入体内可同时抑制乙酰胆碱酯酶(AChE)和丁酰胆碱酯酶(BChE),通过控制胆碱能神经元传导改善AD患者胆碱能介导的记忆、认知功能障碍。成药重酒石酸卡巴拉汀,又名利斯的明、艾斯能,由诺华研发上市,2000年获批进入中国市场,市场应用前景可观。
1 卡巴拉汀合成方法技术演进
本文针对涉及卡巴拉汀整体合成路线的技术分支予以阐述,主要分为两大类:第一类合成方法是利用外消旋体化学拆分,拆分终产物卡巴拉汀外消旋体,或者对卡巴拉汀中间体的外消旋体进行拆分,最终制得左旋的卡巴拉汀;第二类合成方法是不对称合成,主要是先合成出单一构型的(S)-3-[1-(二甲基氨基)乙基]苯酚,即卡巴拉汀的关键中间体,然后与甲基乙基氨基酰氯进行酰化反应,制得左旋的卡巴拉汀。
2 外消旋体化学拆分法合成卡巴拉汀
卡巴拉汀的早期合成路线多是外消旋化学拆分法。1988年诺华公司DE3805744A1[1]申请中首次公开可用于全身透皮给药的游离碱或酸加成盐形式的卡巴拉汀化合物,并最早公开了左旋卡巴拉汀合成路线,后续很多卡巴拉汀拆分合成路线就是在此基础上进行研究和改进的。该专利申请中报道左旋卡巴拉汀是由外消旋体(±)-N-乙基-3-[(1-二甲基氨基)乙基 ]-N-甲基-苯氨基甲酸酯与 (+)-二-O,O’-对甲苯甲酰酒石酸,在甲醇/水混合溶剂中进行拆分得到,其中卡巴拉汀的外消旋体制备方法援引1986年的欧洲申请EP0193926A2。该专利文件中要求保护具有抗胆碱酯酶活性的苯氨基甲酸酯类化合物,其中实施例1制备的化合物即为卡巴拉汀的外消旋体,是由α-羟基苯基乙基二甲基胺与二乙基氨基甲酰氯在乙腈溶剂、NaH存在下进行酯化反应。这种方法的优点是制备步骤少,使用成熟的酯化反应,但所用起始原料难获得,外消旋体制备后拆分成本较高,且使用了危险系数较高的NaH作为缩合剂。
2001年蒋咏文等[2]对外消旋卡巴拉汀的合成路线进行分解,分别合成侧链A(3-[1-(二甲基氨基)乙基]苯酚)和链B(N-甲基-N-乙基氨基酰氯)后,再合成外消旋体,最后拆分得到左旋卡巴拉汀,其路线如图1所示。其中侧链A是以自制间羟基苯乙酮为起始原料与盐酸羟胺在乙醇和氢氧化钠水溶液中反应生成肟,Al-Ni合金存在下还原得到相应胺,再经Eschweiler-Clarke甲基化反应制得3-[1-(二甲基氨基)乙基]苯酚;链B则是套用已知方法得到甲基乙基胺后与光气反应得到甲乙氨基酰氯,同时也记载可由三乙胺与氯甲酸三氯甲酯反应制备甲乙氨基甲酰氯,但该文作者认为光气化反应更利于工业化生产;消旋体卡巴拉汀以3-[1-(二甲基氨基)乙基]苯酚与甲乙氨基酰氯按照已知方法制备,同时合成时试用吡啶作为碱,产率70%左右。该方法优点为原料可自制或易得,但整体路线反应步骤多,尤其是甲基化反应中产物复杂且难以分离,还原时使用的Al-Ni合金容易引入金属离子,增加纯化难度,甲乙氨基甲酰氯制备过程中使用光气,具有危险性,整体路线总收率较低。

图1 蒋咏文等公开的卡巴拉汀合成路线
2007年冯金等[3]在蒋咏文等合成方法基础上,改进性提出在间羟基苯乙酮制备肟步骤中使用弱碱NaHCO3替换NaOH,并将3-羟基苯乙酮肟分离后再投入后续反应,还原胺化步骤是分批加入还原剂Al-Ni,3-(1-氨基乙基)苯酚制备后会由于溶解度问题导致提取困难,在此使用索式提取器提取,3-(1-氨基乙基)苯酚收率达到78%;Eschweiler-Clarke甲基化反应中,通过增加甲基化试剂甲醛和还原剂甲酸用量并延长反应时间,有效提升了中间体3-(1-(二甲基氨基)乙基)苯酚的收率;对于对甲乙氨基甲酰氯合成时,使用毒性较弱的三光气代替光气,满足环保需求,也适宜工业化生产;而3-(1-(二甲基氨基)乙基)苯酚和甲乙氨基甲酰氯酯化得到外消旋卡巴拉汀,再经过拆分并与酒石酸成盐后,总产率达到4.17%。该合成路线虽然一定程度上克服分离和后处理困难的问题,同时避免使用毒性试剂,但反应步骤仍然较多,总产率不高。
2004年印度专利申请IN2004MUM00682A[4]同样报道了以间羟基苯乙酮为原料制备得到3-[1-(二甲基氨基)乙基]苯酚盐酸盐后,与甲乙氨基甲酰氯反应得到消旋体卡巴拉汀,再与DDTA拆分得到左旋卡巴拉汀,其中间羟基苯乙酮制备3-[1-(二甲基氨基)乙基]苯酚盐酸盐采用两种路线,路线A是在间羟基苯乙酮与二甲胺气体在异丙醇钛(IV)、硼氢化钠、IPA/HCl存在下反应,路线B是间羟基苯乙酮与二甲胺气体在Raney Ni/H2存在下反应。
2004年申请WO2004037771A1[5]报道了氮气氛围下,在钛酸四异丙酯存在下,二甲胺与间甲氧基苯乙酮反应后,经硼氢化钠还原得到制备3-[1-(二甲基氨基)乙基]苯甲醚,然后采用氢溴酸试剂脱除甲基制得3-[1-(二甲基氨基)乙基]苯酚,此时对中间体3-[1-(二甲基氨基)乙基]苯酚进行拆分,拆分剂选择廉价易得的S-(+)-樟脑-10-磺酸,得到(S)-3-[1-(二甲基氨基)乙基]苯酚后,与甲乙氨基酰氯反应得到左旋卡巴拉汀,有效降低了N-甲基-乙基氨基酰氯用量成本,最后成盐得到酒石酸卡巴拉汀。2016年昆明源瑞制药有限公司申请CN106565543A[6]也公开以消旋3-(1-(二甲氨基)乙基)苯酚为原料,经S-(+)-樟脑磺酸拆分得到S-3-(1-(二甲氨基)乙基)苯酚后与N-乙基-N-甲基氨基甲酰氯反应得到卡巴拉汀游离碱,最后与L-(+)-酒石酸成盐得到重酒石酸卡巴拉汀。
2008年潘劼[7]对WO2004037771A1报道路线进行研究优化,通过对各步骤条件横向对比试验,使得最终反应总收率达到6.7%;还提出了较好的3-[1-(二甲基氨基)乙基]苯甲醚合成方法是采用Pa/C催化加氢;同时提出对余3-[(1-二甲基胺基)乙基]苯甲醚拆分时,S-(+)-樟脑磺酸、L-(+)-酒石酸和R-磷酸试剂均都不是优良的拆分剂,而L-(+)-酒石酸和R-磷酸试剂不能拆分3-[1-(二甲基氨基)乙基]苯酚,同时印证了S-(+)-樟脑磺酸可以拆分3-[1-(二甲基氨基)乙基]苯酚,拆分后的S-3-(1-(二甲氨基)乙基)苯酚需要重结晶三次满足光学纯度要求。2012年江苏康倍得药业有限公司申请CN103896787A[8]同样提出采用催化常压氢化还原制备[1-(3-甲氧基苯基)乙基]二甲胺,比采用硼氢化钠还原氨化降低了成本和环境污染,催化剂采用雷尼镍、氧化铂或钯碳,未反应的原材料易分离,催化剂也可以回收再利用。
2004年申请WO2005061446A2[9]报道了两条卡巴拉汀合成路线,路线一以间羟基苯乙酮在碳酸钾存在下与甲基乙基氨基甲酰氯反应生成3-O-(N-乙基,甲基)氨基甲酰)苯乙酮,然后用氰基硼氢化钠还原得到相应的醇化合物,后与二甲胺还原氨化生成外消旋卡巴拉汀,用D-DTTA拆分得到卡巴拉汀后与成盐得到酒石酸卡巴拉汀。路线二同样以间羟基苯乙酮为起始原料与N-甲基-N-乙基氨基酰氯反应下生成3-O-(N-乙基,甲基)氨基甲酰)苯乙酮,采用硼氢化钠还原,用三溴化磷进行溴代后再经还原胺化,D-DTTA拆分得到卡巴拉汀。两条路线中,路线二虽然反应步骤多,但各步骤产率较高,所用试剂造成环境污染小,是更优方案。
2006年暨南大学专利申请CN1962624A[10]报道以间羟基苯乙酮与N,N-二甲基甲酰胺通过Leuckart反应一步合成中间体3-[1-(二甲基氨基)乙基]苯酚,采用路易斯酸类催化剂,该方法属于常压反应,工艺难度低,反应收率高。2013年成都医路康医学技术服务有限公司CN103664702A[11]报道类似反应Leuckart反应合成3-(1-(二甲基氨基)乙基)苯酚,其中以间羟基苯乙酮和二甲胺为启示原料。
2007年周黎丽[12]在合成左旋卡巴拉汀时,选择将拆分步骤放在合成开始部分,即3-甲氧基苯乙酮在Raney Ni催化下还原胺化得1-(3-甲氧基苯基)乙胺,就用(S)-扁桃酸拆分对其进行拆分,高回收率得到(S)-1-(3-甲氧基苯基)乙胺后再进行后续合成反应,沿用现有的Eschweiler-Clark甲基化反应得到(S)-[1-(3-甲氧基苯基)乙基]二甲胺,用48%氢溴酸脱甲基制备(S)-3-[1-(二甲基氨基)乙基]苯酚;左旋卡巴拉汀制备时用无水碳酸钾为碱,加入相转移催化剂四丁基溴化铵,最后在丙酮溶剂中成盐得到重酒石酸利斯的明,该方法中创新使用廉价易得的拆分剂(S)-扁桃酸,拆分后(S)-3-[1-(二甲基氨基)乙基]苯酚光学纯度高,工艺总收率达到20.3%,具体路线如图2所示。

图2 周黎丽等公开的卡巴拉汀合成路线
3 卡巴拉汀不对称合成法
不对称合成是有机合成领域的研究热点,该方法虽然工艺简单,但由于多需要使用到手性催化剂导致应用局限性也大,卡巴拉汀不对称合成相比于外消旋体拆分法研究相对较少。
2005年WO2005058804A1[13]报道了先用N-甲基乙胺滴加三光气溶液反应制备得到N-甲基-N-乙基氨基酰氯,再和间羟基苯乙酮反应生成3-O-(N-乙基,甲基)氨基甲酰)苯乙酮。此时以金属铑络合物为手性催化剂,通过不对称还原反应将3-O-(N-乙基,甲基)氨基甲酰)苯乙酮的酮羰基还原为R构型的手性醇,再在甲磺酸酐存在下,与二甲胺进行反应得到构型翻转的左旋卡巴拉汀,最后成盐得到酒石酸卡巴拉汀。该不对称合成方法优点对映体过量(e.e.)值和收率均较高,e.e.值达到95%,总收率55.4%。但由于使用不对称金属铑络合物催化剂,价格昂贵,生产成本高。
2013年无锡佰翱得生物科学有限公司申请CN103304447A[14]针对WO2005058804A1报道的合成路线,改进了手性制备过程中的催化还原试剂,采用(S)-(-)-α,α-二苯基脯氨醇与硼酸三甲酯和硼烷的络合物进行催化手性还原,催化剂可以回收再利用,降低生产成本。
2005年印度专利IN2005MUM00747A[15]公开了由手性原料直接制备左旋卡巴拉汀,可以选择由间羟基苯乙酮与手性甲基苯乙胺原料反应制得亚胺中间体后,得到手性(S)-3-(1-甲基氨基)乙基)苯酚,再与甲基乙基甲酰氯制得卡巴拉汀,或由带有N-取代基的手性中间体与甲基乙基甲酰氯反应后,再脱除N-取代基得到左旋卡巴拉汀。2007年的国际申请WO2008037433A1[16]也公开了以手性3-(1-二甲基氨乙基)苯酚与二(4-硝基)苯基碳酸酯反应后,手性中间体再与甲基乙基甲酰氯反应得到卡巴拉汀。
2009年上海医药工业研究院两份专利申请CN101481333A[17]和CN101481334A[18]均采用已知手性化合物3-((S)-1-((S)-1-苯乙胺基)乙基)苯基乙基(甲基)氨基甲酸酯为起始原料手性不对称合成卡巴拉汀。其中CN101481333A公开了3-((S)-1-((S)-1-苯乙胺基)乙基)苯基乙基(甲基)氨基甲酸酯先经胺甲基化、催化氢化脱乙苯基保护、再在多聚甲醛和甲酸作为甲基化试剂的情况下反应得到左旋卡巴拉汀;CN101481334A中3-((S)-1-((S)-1-苯乙胺基)乙基)苯基乙基(甲基)氨基甲酸酯在钯碳等催化剂下催化氢化脱除乙苯基得到手性中间体,再经胺甲基化得到左旋卡巴拉汀。同时均公开了起始原料是由3-乙酰苯基乙基(甲基)氨基甲酸酯和手性胺在钛四异丙酯催化下制备得到。
2012年浙江九州药物科技有限公司申请CN103193679A[19]报道了卡巴拉汀中间体R-N-乙基-N-甲基氨基甲酸-3-(1-羟乙基)苯酯的制备,以N-乙基-N-甲基氨基甲酸酯-3-乙酰基苯酯为原料,采用金属Ir络合物作为手性催化剂,所用手性催化剂用量低,同时利用氢气替代现有技术中的甲酸作为氢源,原子经济学高。
2009年 Juan Mangas-Sánchez等[20]报道了以酶解法制备卡巴拉汀手性中间体原料的合成路线,其中在温和的反应条件下直接化学酶合成对映体纯的卡巴拉汀,其中Candida antarctica lipase B对相应的(R)-醇或胺进行立体选择性乙酰化后,再制备得到手性中间体(S)-5,如图3所示。
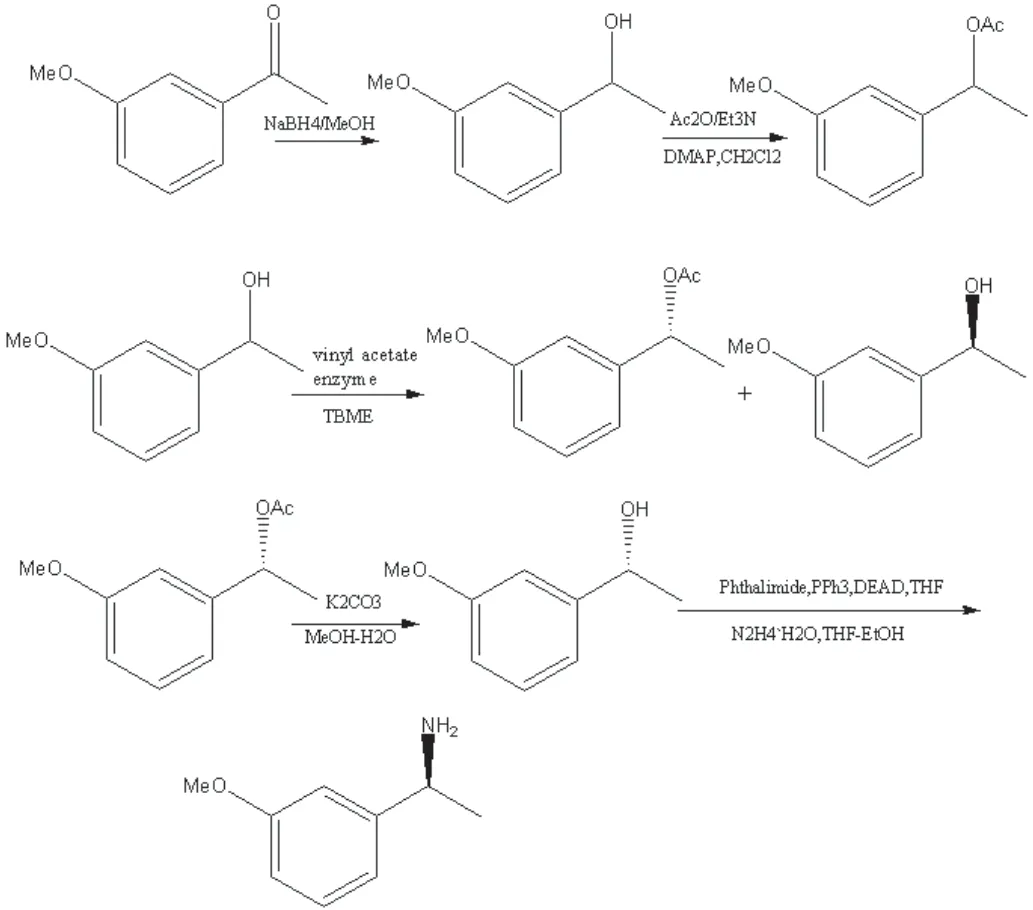
图3 Juan Mangas-Sánchez等公开的卡巴拉汀手性中间体合成路线
在2011年全国药物化学学术会议上郑州大学王姣等[21]发表关于卡巴拉汀的生物化学合成方法,以3’-间羟基苯乙酮为原料,与甲基乙基甲酰氯进行酯化反应,经硼氢化钠还原为消旋仲醇3,然后以乙酯异丙烯酯作为酰基供体对仲醇3进行动力学拆分,该课题组由土壤中筛选出的脂肪酶菌种WJ-3,将其作为催化剂用于制备(R)-4,提出了新的不对称合成生物催化试剂。
2017年Davide Brenna等[22]报道以亚胺化合物为原料,不对称合成卡巴拉汀的方法,该方法采用无金属非对映选择性还原亚胺并随后去除手性助剂,其所用还原剂廉价且无毒,非手性路易斯碱(如 N,N-二甲基甲酰胺)也易得,如图4所示。
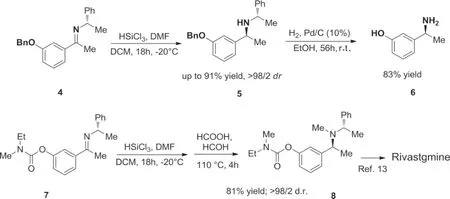
图4 Davide Brenna等公开的卡巴拉汀合成路线
2018年山东师范大学等申请CN108822000A[23]报道了基于Ir络合物催化还原胺化合成(S)-卡巴拉汀的方法:化合物N-甲基乙基(3-乙酰基苯基)氨基甲酸酯与二苯甲胺在Ir络合物催化下得到手性中间体化合物,再加入金属脱苄催化剂得到(S)-卡巴拉汀,产物光学纯度高。
4 结语
目前,卡巴拉汀的市场价格昂贵,且以进口为主,相比于其他治疗阿尔兹海默症的药物,国内市场竞争相对缓和。通过对卡巴拉汀合成技术分支的梳理,为今后寻找更好的合成和拆分方法,提供合适的背景供知识储备,引导国内研究人员寻找合适的突破方向,进而推动创新。
猜你喜欢
安徽农学通报(2022年6期)2022-04-07
文萃报·周五版(2021年42期)2021-10-28
家庭百事通·健康一点通(2020年11期)2020-11-30
师道·教研(2019年10期)2019-11-21
伙伴(2019年6期)2019-07-19
学校教育研究(2017年28期)2017-10-21
科技创新与应用(2017年20期)2017-07-15
家庭用药(2016年8期)2016-05-14
中外文摘(2015年6期)2015-11-22
读写算·小学低年级(2015年9期)2015-09-18
