
NF1致椎体骨质疏松并脊柱畸形的基础研究
2022-10-18 05:19李鹏飞周林贾楠张为何玉秀王金星
中国骨质疏松杂志 2022年9期
李鹏飞 周林 贾楠 张为 何玉秀* 王金星
1. 河北师范大学体育学博士后流动站,河北 石家庄 050024 2. 河北医科大学附属哈励逊国际和平医院骨科中心,河北 衡水 053000 3. 河北医科大学第三医院脊柱科,河北 石家庄 050051
NF1脊柱营养不良性改变包括椎体骨量减少、局部骨质疏松、椎体后缘扇形变、重度椎体旋转、楔形变、高度丢失、椎管扩大、神经根管扩大、椎弓根间距增宽、变异、椎旁肿块生长、横突纺锤形变和肋骨带状改变并扭曲旋转[1-2]。这些变化主要由NF1产生局部活性物质及激素诱发。由于缺少相关动物模型,NF1患者椎体骨质疏松并脊柱畸形的病理学研究进展缓慢[3]。营养不良性椎体骨质疏松和脊柱畸形等病理改变是由于原发性椎体骨骼发育不良?还是椎间孔或邻近组织内神经纤维瘤继发病变?目前仍存有争议[4]。部分NF1基因缺失小鼠模型不能充分用于骨病研究,是由于心内膜过度增殖和过早凋亡使心脏发育中断导致胚胎死亡[5]。虽然部分NF1鼠类模型可显现一定程度骨骼疾病,且类似于人类NF1患者;然而没有相关文献报道此类模型出现椎体骨量减少及脊柱畸形,即不能复现人类NF1患者主要骨骼缺陷[6]。本课题应用不同基因型NF1小鼠模型实验,明确NF1致局部骨量减少并伴随多发性脊柱缺陷的病因,并解释其原发性与继发性改变,观察Nf1flox/-;Col2.3Cre+基因型小鼠脊柱骨骼特征,为深入研究NF1致椎体骨质疏松及脊柱畸形的机制奠定基础。
1 材料和方法
1.1 材料
WT、Nf1+/-基因型小鼠源自麻省理工学院(Cambridge, MA, US)。Nf1flox/flox基因型小鼠由德克萨斯大学西南医学中心提供。培育Col2.3cre转基因小鼠。将成年Col2.3cre转基因小鼠与ROSA26小鼠杂交。PCR检测各型小鼠基因,确认制备基因型为Nf1flox/flox;Col2.3Cre+、Nf1flox/-;Col2.3Cre+小鼠。外周双能X射线吸收测定平台(peripheral dual-energy X-ray absortiometry, pDXA,PIXImus II; GE-Lunar Corp., Madison, WI),外周定量计算机断层扫描平台(pQCT, Stratec Medizintechnik GmbH, Pforzheim, Germany),异氟醚(IsoFlo; Abbott Laboratories, North Chicago, IL),Biopix系统(Bioptics, Inc. Tucson, AZ),Mtestw R,Image J (NIH, Bethesda, MD, US)等,所有研究均符合动物实验伦理学要求。
1.2 方法
1.2.1X射线摄影及骨密度检测:X线数字摄影技术拍摄正位全长脊柱X线片。外周双能X射线吸收测定法观察不同基因型小鼠骨量及骨密度(bone mineral density,BMD)。成年小鼠吸入麻醉后进行头部之外的全身扫描,主要观测中轴骨中央50 %及腰椎区域。
1.2.2外周定量计算机断层扫描:将第五腰椎(L5)置于外周定量计算机断层扫描平台。沿L5椎弓根下缘横截面扫描,观察中央椎体区域。测量皮质骨的体积骨密度(mg/cm3)。计算椎体及椎管横截面积比值。
1.2.3成骨样细胞分离:首先培养成骨样细胞[7-9]:1月龄小鼠处死后去除肌肉结缔组织,分离椎骨。收集腰椎样本;恒温37 ℃旋转摇动40 min,应用III型胶原酶(0.1 mg/mL)和2.5 %胰蛋白酶分解组织并提取骨髓细胞。培养基加入10 % FBS,冲洗游离细胞;细胞悬液通过70 μm过滤器筛选去除聚集体。锥蓝染色测定阴性细胞数量。
1.2.4生物力学测试:解剖L5椎体,去除软骨终板及椎间盘组织并暴露骨面。生物力学测试仪进行椎体压缩测试。速率设定20 mm/min。峰值负载为椎体骨折前所承受最大载荷,同时测量椎体横截面积。峰值压强计算为:峰值压强=峰值负载/椎体横截面积。
1.2.5成骨细胞分化实验:观察Nf1flox/-;Col2.3Cre+小鼠腰椎骨髓中成骨母细胞数量;成骨细胞集落形成单位。检测成骨细胞分化采用茜素红染色和von Kossa染色;并测定钙盐沉积。饲养1月后处死小鼠,取L1~L5椎体,收集骨髓单核细胞。采用密度梯度离心法将研碎后椎骨和骨髓细胞分离[10-12]。分组后采用成骨细胞分化培养基培养,每组含2×106/mL骨髓单核细胞。两周后进行碱性磷酸酶及茜素红染色。Image J软件计数细胞。Von-kossa银法染色并观察骨髓细胞成骨分化,记录细胞形成矿化结节情况。
1.2.6组织学检查:将6月龄小鼠处死。解剖分离腰椎浸泡于4 %甲醛溶液。随后置于含10 %乙二胺四乙酸和4 %甲醛溶液中脱钙4 d。10 %甲醛固定、乙醇分级脱水后石蜡包埋,制作5 μm切片。获得L5椎体纵断面切片,进行抗酒石酸酸性磷酸酶(TRAP)和MacNeal染色[13]。进行破骨细胞和成骨细胞计量分析。
1.3 统计学处理

2 结果
2.1 Nf1 flox/-;Col2.3Cre+小鼠椎体发育不良性椎体高度减小
四组比较,虽然各基因型小鼠腰椎平均高度比较差异无统计学意义(见图1A),但是Nf1flox/-;Col2.3Cre+小鼠L5椎体平均高度低于WT、Nf1+/-、Nf1flox/flox;Col2.3Cre+小鼠(见图1B、1C、P<0.05)。差异有统计学意义。表明Nf1flox/-;Col2.3Cre+小鼠营养不良性椎体高度减小与NF1致脊柱畸形特点相符合。
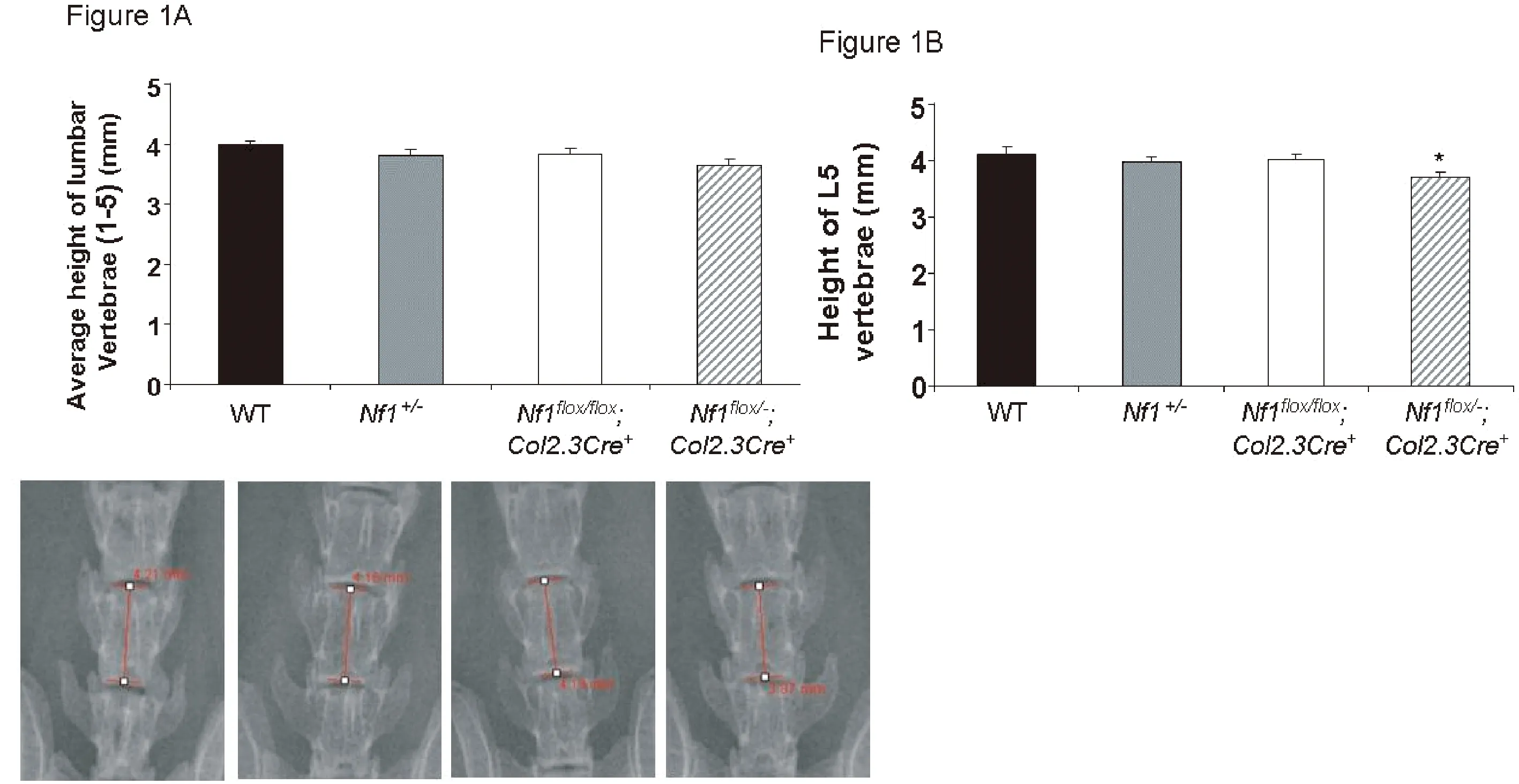
注:A:L1~L5椎体平均高度。B:L5椎体平均高度。C:L5椎体X光影像学测量。图1 Nf1flox/-;Col2.3Cre+小鼠椎体高度下降Fig.1 Vertebral average height of Nf1flox/-;Col2.3Cre+ mice decreased
2.2 Nf1flox/-;Col2.3Cre+小鼠L1~ L5椎体骨密度降低
首先测量4组小鼠完整脊柱骨密度。然后对L1~L5分别测量(见图2A)。结果示4组小鼠脊柱中轴骨整体骨密度比较差异无统计学意义(见图2B)。但腰椎骨密度比较差异有统计学意义。其中,Nf1flox/-;Col2.3Cre+小鼠L1~ L5区域骨密度明显降低(见图2C,*P<0.05)。
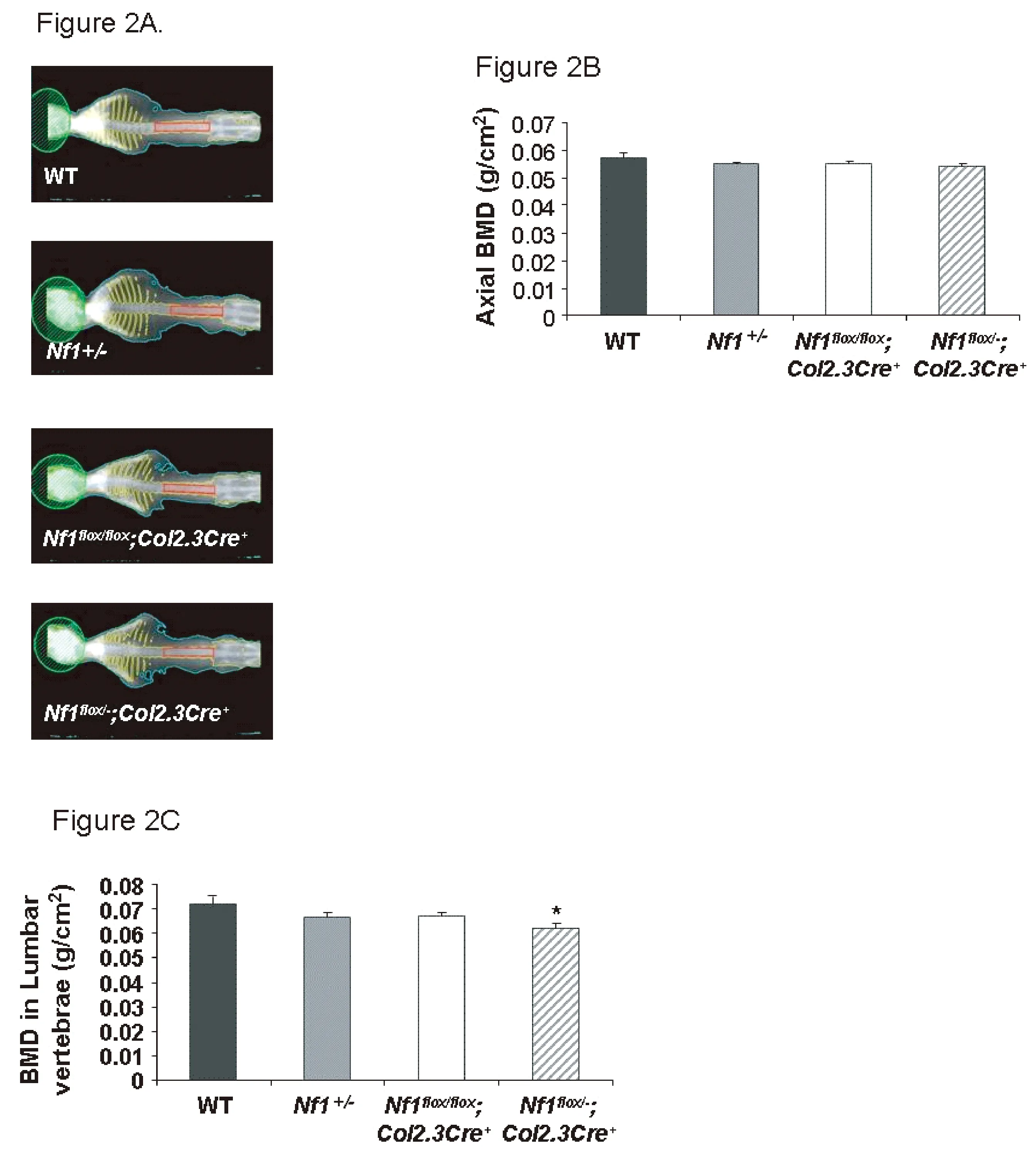
注:A:小鼠中轴骨的骨密度,红色区域为L1~ L5。B:中轴骨总体骨密度。C:L1~ L5骨密度。图2 Nf1flox/-;Col2.3Cre+小鼠L1~ L5骨密度减小Fig.2 L1-L5 BMD of Nf1flox/-;Col2.3Cre+ mice decreased
2.3 Nf1flox/-;Col2.3Cre+小鼠L5椎体皮质骨量和骨密度均降低
Nf1+/-与Nf1flox/flox;Col2.3Cre+小鼠比较差异无统计学意义,然而Nf1flox/-;Col2.3Cre+小鼠L5椎体皮质骨密度明显降低(见图3A、3B,**P<0.05)。同时,不仅Nf1+/-与Nf1flox/flox;Col2.3Cre+组与对照组比较L5椎体皮质骨量均下降,且Nf1flox/-;Col2.3Cre+小鼠皮质骨面积下降更显著(见图3C,**P<0.01)。4组椎体面积比较差异无统计学意义(见图3D、3E)。但对比4组C/V值发现,Nf1+/-、Nf1flox/flox;Col2.3Cre+、Nf1flox/-;Col2.3Cre+组显著大于对照组(见图3F,*P<0.01)。组间比较,Nf1flox/-;Col2.3Cre+组C/V值显著大于Nf1+/-、Nf1flox/flox;Col2.3Cre+组(**P<0.01)。因此,Nf1flox/-;Col2.3Cre+小鼠模型椎管扩大增宽符合NF1患者椎管结构特点。
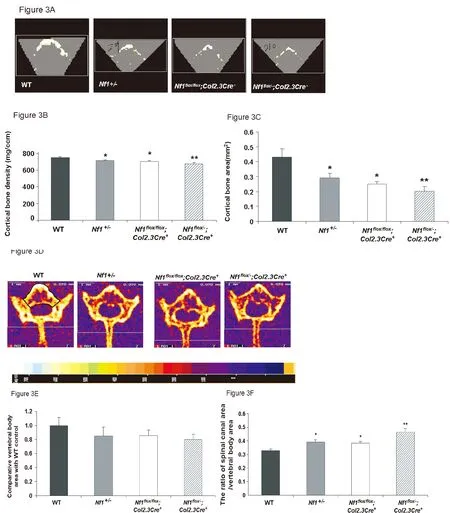
注:A:L5椎体断层扫描。B:L5椎弓根下缘皮质骨密度。C:L5椎体皮质骨面积。D:L5横截面椎管及椎体结构。E:椎体面积。F:椎管面积与椎体面积比值。图3 Nf1flox/-;Col2.3Cre+小鼠皮质骨面积和骨密度均降低。Fig.3 The cortical bone area and BMD of Nf1flox/-;Col2.3Cre+ mice decreased
2.4 Nf1flox/-;Col2.3Cre+小鼠椎体承重能力降低
对4组小鼠L5椎体增大负载至椎体破裂,观察受力峰值分布曲线、记录最大压力负荷(见图4A),发现:Nf1+/-组最大压力负荷为25 N、Nf1flox/flox;Col2.3Cre+组为24 N、Nf1flox/-;Col2.3Cre+组为20 N,明显小于对照组35 N(见图4B,*P<0.05)。组间比较,Nf1flox/-;Col2.3Cre+组明显小于Nf1+/-、Nf1flox/flox;Col2.3Cre+组(见图4B,**P<0.01)。
承受最大压强比较:实验组明显小于对照组(见图4C,*P<0.05)。组间比较,Nf1flox/-;Col2.3Cre+组显著小于Nf1+/-、Nf1flox/flox;Col2.3Cre+组(见图4C,**P<0.01)。基于上述发现,Nf1flox/-;Col2.3Cre+组承受最大负荷及最大压强均明显减弱。此结果与之前实验中Nf1flox/-;Col2.3Cre+小鼠L5皮质骨量和骨密度均显著降低之间存在必然联系。

注:A:载荷峰值分布曲线图。B:最大压力负荷。C:最大压强。图4 L5椎体峰值压力与压强比较Fig.4 Comparison between peak load and stress on L5
2.5 L5椎体组织学检验
Nf1+/-、Nf1flox/-;Col2.3Cre+组L5上关节突骨陷窝面积显著大于Nf1flox/flox;Col2.3Cre+组和对照组(见图5A,*P<0.01)。Nf1+/-、Nf1flox/-;Col2.3Cre+组破骨细胞数量明显多于Nf1flox/flox;Col2.3Cre+组和对照组(见图5B,*P<0.01);组内比较Nf1flox/-;Col2.3Cre+组破骨细胞数量明显多于Nf1+/-组(见图5B,**P<0.01)。Nf1flox/-;Col2.3Cre+组成骨细胞数量及密度显著低于对照组、Nf1+/-组及Nf1flox/flox;Col2.3Cre+组(见图5C、5 D,**P<0.01)。

注:A:TRAP染色(20×)。B:破骨细胞区域面积量化比较。C:MacNeal染色(20×)。D:成骨细胞定量分析。图5 Nf1flox/-;Col2.3Cre+组破骨细胞区域增加,成骨细胞数量减少Fig.5 The area of osteoclasts in Nf1flox/-;Col2.3Cre+ group increased and the number of osteoblasts decreased
2.6 NF1缺失抑制体外培养成骨细胞分化
与对照组相比,Nf1flox/-;Col2.3Cre+小鼠骨髓单核细胞体外培养获得成骨细胞集落形成单位能力显著降低(见图6A,**P<0.01)。对照组茜素红染色强阳性,而Nf1+/-、Nf1flox/flox;Col2.3Cre+、Nf1flox/-;Col2.3Cre+组茜素红染色减弱(见图6B,**P<0.01);并且矿化骨结节数量显著减少(见图6C,P<0.01)。表明Nf1+/-和Nf1-/-椎体内前成骨细胞和成骨细胞分化及矿物质沉积功能受损。而Nf1flox/-;Col2.3Cre+组功能最弱(***P<0.01)。

注:A:碱性磷酸酶染色。B:茜素红染色。C:Von Kossa染色。图6 成骨细胞分化形成抑制实验Fig.6 Inhibition of differentiation and formation of osteoblasts
3 讨论
NF1是一种常见的常染色体显性遗传病,由NF1抑癌基因突变引起[14]。脊柱骨量丢失及结构畸形是NF1患者常见骨骼病变表现[15]。目前,由于缺乏合适的动物模型,NF1发生椎体骨骼异常机制仍不清楚[16-17]。本研究利用基因杂合法,建立Nf1flox/-;Col2.3Cre+小鼠模型。实验发现此NF1小鼠脊椎节段较短,椎体骨皮质和骨量均明显减少;椎体峰值压力和峰值负荷均显著降低;并复现出NF1患者椎管扩大的腰椎病变特征;组织学分析显示小鼠椎体破骨细胞形成增加,成骨细胞分化功能减弱。总之,Nf1flox/-;Col2.3Cre+小鼠表现出多种腰椎结构和功能异常,再现NF1患者脊柱营养不良性改变。此NF1小鼠亦提供了深入了解NF1患者椎体骨质疏松及脊柱脊髓功能异常的动物模型,为深入研究NF1的病理机制奠定基础。
目前,NF1致椎体骨骼发育异常的机制尚不完全清楚[18]。临床上,NF1致椎体骨质疏松和脊柱畸形进展的严重程度,均与椎体特征性形态发生改变有关[19]。而且NF1致脊柱畸形是原发性骨骼发育缺陷,还是源自邻近椎体旁肿瘤影响,目前仍存在争议[20-22]。通过本研究发现Nf1flox/-;Col2.3Cre+动物模型出现椎体骨量明显减少、破骨细胞形成增加及成骨细胞数量减少,表明脊柱存在原发性椎体骨骼发育缺陷[23]。
哺乳动物骨骼发育和重塑是活跃且持续的过程,需要破骨细胞和成骨细胞互相调整来维持体内生长平衡[24-26]。营养不良性椎体骨质疏松和脊柱畸形等病理改变是由于原发性椎体骨骼发育不良?还是椎间孔或邻近组织内神经纤维瘤的继发性反应?结合此项研究,课题组认为是NF1导致了破骨细胞形成增加和成骨细胞生成减少。这些细胞系原发性病理缺陷可能导致椎体骨量减少和脊柱畸形快速进展[27]。亦可解释NF1临床特异性表现:包括NF1患者脊柱矫形术后内固定并发症及脊柱融合术后假性关节病发生率均较高[28-30]。
本研究亦存在局限性,实验中所建立NF1小鼠动物模型为四足类,脊柱不承担纵向压力,同时不需要维持脊柱平衡的轴向载荷。但人类两足运动,脊柱为主要载荷部位。由于两足类脊柱生物力学情况与四足类不同,因此四足动物模型不能完全显示两足类脊柱畸形特征。包括NF1患者特征性扇贝样改变及脊柱后凸畸形。尽管有条件限制,Nf1flox/-;Col2.3Cre+小鼠与对照组相比,仍然出现脊柱椎体皮质骨发育异常、骨量减少情况,以及腰5椎管结构增宽并发育异常。目前课题组亦积极攻关,拟建立NF1致椎体骨量减少并脊柱侧弯的双足类动物模型。
综上所述,本研究证据表明NF1中存在原发性成骨细胞以及破骨细胞功能缺陷,可能导致NF1患者出现发育营养不良性骨质疏松以及多发性脊柱畸形。该实验为进一步研究NF1椎体原发性骨质疏松与脊柱形态学改变的机制进行铺垫,为深入了解及治疗NF1患者骨质疏松及脊柱畸形提供科学依据。
猜你喜欢
健康研究(2022年5期)2022-11-07
中国兽医学报(2022年4期)2022-06-17
中国典型病例大全(2021年7期)2021-08-02
中国药学药品知识仓库(2021年18期)2021-02-28
家庭医学(2020年12期)2020-01-20
中外医学研究(2019年18期)2019-08-19
中国医药导报(2017年6期)2017-04-06
中国中药杂志(2017年2期)2017-03-25
华西口腔医学杂志(2014年6期)2014-10-21
上海医药(2014年9期)2014-06-27
