
Liver-specific drug delivery platforms: Applications for the treatment of alcohol-associated liver disease
2022-10-18 01:49:00JeffreyBarrWarnerStevenCorriganGuenthnerJosiahEverettHardestyCraigJamesMcClairDennisRayWarnerIrinaAndreyevnaKirpich
World Journal of Gastroenterology 2022年36期
Jeffrey Barr Warner, Steven Corrigan Guenthner, Josiah Everett Hardesty, Craig James McClair,Dennis Ray Warner,Irina Andreyevna Kirpich
Abstract Alcohol-associated liver disease (ALD) is a common chronic liver disease and major contributor to liver disease-related deaths worldwide. Despite its prevalence, there are few effective pharmacological options for the severe stages of this disease. While much pre-clinical research attention is paid to drug development in ALD, many of these experimental therapeutics have limitations such as poor pharmacokinetics, poor efficacy, or off-target side effects due to systemic administration. One means of addressing these limitations is through liver-targeted drug delivery, which can be accomplished with different platforms including liposomes, polymeric nanoparticles, exosomes, bacteria, and adenoassociated viruses, among others. These platforms allow drugs to target the liver passively or actively, thereby reducing systemic circulation and increasing the‘effective dose’ in the liver. While many studies, some clinical, have applied targeted delivery systems to other liver diseases such as viral hepatitis or hepatocellular carcinoma, only few have investigated their efficacy in ALD. This review provides basic information on these liver-targeting drug delivery platforms, including their benefits and limitations, and summarizes the current research efforts to apply them to the treatment of ALD in rodent models. We also discuss gaps in knowledge in the field, which when addressed, may help to increase the efficacy of novel therapies and better translate them to humans.
Key Words: Liver targeted delivery; Nanoparticles; Liposomes; Polymeric nanoparticles; Precision medicine;Alcohol associated liver disease
INTRODUCTION
Pathogenesis and pharmacological management of alcohol-associated liver disease
Alcohol-associated liver disease (ALD) is a common chronic liver disease and contributes to the global healthcare burden caused by excess alcohol consumption, which is defined as more than 1 or 2 standard drinks of alcohol per day for females and males, respectively. Globally, nearly half of liver cirrhosis deaths are attributed to alcohol abuse[1]. The pathogenesis of ALD follows a well-described pattern of disease stages beginning with simple liver steatosis progressing to steatohepatitis (steatosis with inflammation), cirrhosis (advanced liver fibrosis), and in some severe cases, hepatocellular carcinoma (HCC)[2] (Figure 1A). In individuals who chronically consume alcohol, binge-drinking episodes may cause acute alcohol-associated hepatitis (AH), a life-threatening condition with high short-term mortality due to infection, severe inflammation, and multi-organ failure[3]. The pathophysiology of ALD is multifactorial and involves a variety of effects of alcohol on multiple organs, including the liver and the gut (Figure 1B). For example, alcohol-induced intestinal permeability and subsequent translocation of gut bacteria and bacteria-derived products into the portal circulation may contribute to inflammation,hepatic stellate cell (HSC) activation, and fibrosis in the liver. Further, direct effects of alcohol on the liver may result in dysregulated lipid signaling, hepatocyte cell death, and production of reactive oxygen species leading to steatosis as well as further inflammation, fibrosis, and ultimately liver cancer(these concepts have been reviewed in detail previously[2]). Most patients with early-to-mid-stage ALD(i.e., hepatic steatosis or mild steatohepatitis) are asymptomatic, therefore a diagnosis of ALD is often not made until later stages of the disease. In those individuals where a diagnosis is made, abstinence and nutrition are key, and indeed, some stages of the disease (e.g., steatosis) are reversible upon alcohol cessation. Limited pharmacological options exist for patients with alcohol-related late-stage liver disease(e.g., cirrhosis or AH), including prednisolone (a corticosteroid) and pentoxifylline (a phosphodiesterase inhibitor used in patients for which corticosteroids are contraindicated or not effective), but importantly,these drugs only reduce short-term mortality[4,5]. There is much research attention being given to drug development in ALD using animal models. These therapies target various pathogenic mechanisms in ALD including oxidative stress (e.g., S-adenosylmethionine, betaine, natural antioxidants), inflammation[e.g.,anti-tumor necrosis factor (TNF) therapy, interleukin (IL)-22, glucocorticoids, steroids, IL-1R inhibitors, granulocyte-colony stimulating factor], fibrosis (e.g., transforming growth factor-β inhibitors,phosphodiesterase inhibitors, PPAR agonists), gut barrier dysfunction and microbial dysbiosis (e.g.,probiotics and antibiotics), and other processes (these drugs and others are thoroughly reviewed in[6]).
Overview of liver-specific drug delivery systems

Figure 1 Spectrum and pathophysiology of alcohol-associated liver disease. A: Schematic diagram describing the spectrum of disease stages in alcohol-associated liver disease (ALD). Percentages represent proportion of chronic drinkers who progress to the indicated disease stage; B: Schematic diagram depicting the pathophysiology of ALD. Ethanol affects both the gut and liver to induce changes in lipid metabolism, generation of reactive oxygen species and hepatocyte cell death, gut permeability, and downstream consequences including inflammation, hepatic stellate cell activation, fibrosis, DNA damage, and carcinogenesis. ALD: Alcohol-associated liver disease; ROS: Reactive oxygen species; HSC: Hepatic stellate cell.
While much pre-clinical research attention is given to new drug development for liver diseases,including ALD, many experimental therapeutics relying on systemic drug administration suffer from drawbacks including poor pharmacokinetics or a low margin of safety due to off-target effects in other organs. An example of an early attempt to address some of these drawbacks is covalent conjugation of polyethylene glycol (PEG) to drug molecules (termed ‘PEGylation’), a strategy that has been used for many years to lengthen half-life, improve water solubility, and decrease immunogenicity[7]. For instance, PEGylated interferon-α has been the first line treatment for chronic hepatitis B since 2005[8].However, since that time, advances in nanomedicine have produced numerous liver-specific drug delivery platforms based on lipid vesicles, inorganic nanoparticles, and biological systems which allow:(1) Improved pharmacokinetics for drugs with poor solubility, low bioavailability, rapid metabolism,etc.; (2) Reduced systemic side effects by delivering drugs to the liver while avoiding other organs; and(3) Improved efficacy of drugs intended to act in the liver by increasing the ‘effective dose’.
There are several types of liver-targeting drug delivery platforms which can be broadly categorized by their composition, including: lipid-based particles (e.g., micelles, liposomes, and exosomes,Figure 2A), non-lipid-based particles [e.g., polymeric nanoparticles (PNPs), metallic nanoparticles, and ceramic nanoparticles, Figure 2B], and bacterial and viral platforms (e.g., bioengineered bacteria and adeno-associated viruses, Figure 2C). These systems are either synthetic or derived from living systems,and have distinct advantages and disadvantages based on their efficacy, pharmacokinetics, and side effects (summarized in Table 1). Briefly, lipid-based particles are composed of endogenous lipids which keep the risk of immunogenicity and toxicity low. Metallic, ceramic, and some PNPs are nonbiodegradable and sometimes cytotoxic, but can be modified to reduce toxicity and have additional uses in medical imaging and diagnostics[9]. Bacterial and viral drug delivery platforms benefit from the natural tropism of certain bacteria or viruses for a particular organ or niche but are also potentially immunogenic. These liver-targeting approaches have been used for the treatment of various liver diseases including HCC (e.g., liposomal, PEGylated, or PNP-encapsulated anti-cancer compounds[10-12]), viral hepatitis (e.g., metal nanoparticles[13] and PEGylated interferon[8]), and liver fibrosis (e.g.,liposomal vitamin A[14]) with some reaching full FDA approval (e.g., Pegasys, Miriplatin, and others)[15].
Biodistribution of liver-targeted drug delivery platforms

Table 1 Summary of liver-specific drug delivery platforms, including molecular composition, potential modifications, benefits, and limitations
The biodistribution of liver-specific drug delivery platforms in the body after systemic administration is based on the physical properties of the particle. For example, before reaching target liver cells, many particles may be opsonized by binding plasma proteins (e.g., albumin, apolipoproteins, antibodies,complement component proteins) and cleared by the reticuloendothelial system (RES) of the liver and spleen, including by LSECs, particularly if the particles are greater than 200 nm in diameter or carry a negative charge[18]. Particle modifications such as PEGylation help avoid RES surveillance by preventing plasma protein binding, thereby improvingin vivohalf-life. Stealth liposomes, for example,are PEGylated phospholipid particles commonly used to improve the pharmacokinetics of a drug with a short half-life[19]. Particles which avoid RES clearance and have favorable size and charge can pass through the liver sinusoidal fenestrae, which are approximately 100-150 nm in diameter[20], to access HSCs in the space of Disse and the liver parenchyma. Accordingly, particles must have roughly the same or smaller diameter than these fenestrae and carry a charge which is not excessively positive or negative, as high charge magnitude is associated with increased plasma clearance[21]. Controlling these physical properties to allow accumulation of particles in the liver is called passive liver targeting,whereas active liver targeting relies on conjugation of a “homing” ligand whose receptor is expressed in the target organ, and in particular, the specific target cell type. For example, carbohydrate receptors such as the asialoglycoprotein receptor can be targeted to deliver therapeutics to hepatocytes with ligands including galactose, lactose, pullulan, and others (more information regarding active targeting has been reviewed by Kanget al[22]).
LIVER-SPECIFIC DRUG DELIVERY: IMPLICATIONS FOR ALD

Figure 2 Graphical representation of targeted drug delivery platforms. A: Lipid-based particles, including micelles, liposomes, and exosomes; B: Nonlipid-based particles, including polymeric nanospheres and nanocapsules, metallic nanoparticles, and ceramic nanoparticles; C: Bioengineered bacteria and adenoassociated virus serotype 8. Graphics are not drawn to scale. miRNA: MicroRNA; AAV8: Adeno-associated virus serotype 8.
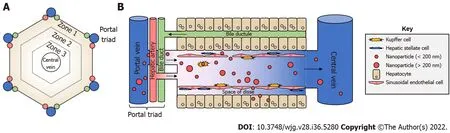
Figure 3 Lobular liver architecture and biodistribution of nanoparticles. A: Top-down view of a liver lobule. Portal triads, consisting of a portal vein,hepatic artery, and bile duct, surround a central vein in a hexagonal shape. Concentric hexagons designate zones 1-3 moving from the outside to the inside. Portal and arterial blood flows from the triads toward the central vein, whereas bile travels the opposite direction; B: Side view. With the portal triad on the left, portal blood brings nanoparticles from the digestive tract to the liver sinusoids where they can interact with Kupffer cells, liver sinusoidal endothelial cells, and others.Nanoparticles of sufficiently small size can pass through the liver fenestrae formed by liver sinusoidal endothelial cells to access the Space of Disse, and subsequently, hepatocytes. Images are not drawn to scale.
The goal of this review is to summarize pre-clinical research efforts which apply liver-specific drug delivery platforms in various rodent models to prevent or treat ALD, as well as to further discuss the drug delivery systems themselves, which include liposomes, exosomes, PNPs, bacteria, and adenoassociated viruses. To this end, we searched the PubMed (https://pubmed.ncbi.nlm.nih.gov), Google Scholar (https://scholar.google.com), and Web of Science (https://www.webofscience.com/wos/wosc c/basic-search) databases for studies published up to June 1, 2022 using a combination of text keywords“alcohol liver disease” and the following: “liposome(s)”, “liposomal”, “nanoparticle(s)”, “nanoformulated”, “polymersome(s)”, “polymeric nanoparticle(s)”, “micelle(s)”, “exosome(s)”, “AAV”,“adenovirus”, “adeno-associated virus”, and “bioengineered bacteria”. Our search strategy identified 846 unique results, which were screened individually by title and abstract and were included based on relevance to liver-targeted drug delivery in ALD. Studies were not excluded based on date of publication, model organism, funding source, or drug delivery platform used. Based on these criteria, 16 studies were included, and then categorized by drug delivery platform (n= 7 studies related to liposomes,n= 2 related to exosomes,n= 5 related to PNPs, andn= 2 related to bacterial or viral systems). A graphical summary of the search strategy and study categorization can be found in Figure 4.The 16 key studies are described in detail in Table 2. The reader is encouraged to refer to this table for information such as the platform employed, the cargo molecule(s), the physical characterization of the particles used (if provided), and the animal model of ALD used, among other information.

Table 2 Summary of studies employing a liver-specific drug delivery platform in animal models of alcohol-associated liver disease

Passive targeting denotes a strategy wherein the physical properties of a particle are modified to target the liver, and active targeting denotes a strategy wherein a particle targets the liver through a ligand/receptor interaction.Treatment paradigm denotes models wherein the drug is administered after liver injury has been established (e.g., half-way through the model, at the end of the model, etc.), whereas prevention paradigm denotes models wherein the drug is administered for the entire duration of the model. Changes in results/mechanisms columns are in liver unless otherwise stated. AAV8: Adeno-associated Virus Serotype 8; ADH1: Alcohol dehydrogenase 1; AhR: Aryl hydrocarbon receptor; AKT: Protein kinase B; ALP: Alkaline phosphatase; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; ATF3: Activating transcription factor 3; cAMP: Cyclic adenosine monophosphate; CCl4:Carbon tetrachloride; CCR2: C-C motif chemokine receptor 2; CHOP: C/EBP homologous protein; COX2: Cyclooxygenase 2; DCF: Dichlorodihydrofluorescein; DHE: Dihydroethidium; EE%: Encapsulation efficiency percent; EGF:Epidermal growth factor; EGFR: Epidermal growth factor receptor; ER: Endoplasmic reticulum; GPx: Glutathione peroxidase; GSH: Glutathione; HDL: High density lipoprotein; HSC: Hepatic stellate cell; i.p.: Intraperitoneal injection;i.v.: Intravenous injection; IL: Interleukin ; iNOS: Inducible nitric oxide synthase; LDL: Low density lipoprotein; LGG: Lactobacillus rhamnosus GG; LPS: Lipopolysaccharide; MCP1: Monocyte chemoattractant protein 1; MDA:Malondialdehyde; miR: Micro-RNA; MMPs: Matrix metalloproteinases; MPO: Myeloperoxidase; mTOR: Mechanistic target of rapamycin; N/A: Not applicable; N/P: Not provided; NRF2: Nuclear factor erythroid 2-related factor 2;ORO: Oil red O; P-AMPKα: Phospho-AMP-activated protein kinase alpha; PCX: Polycationic CXCR4 antagonists; PEG: Polyethylene glycol; PG: Propylene glycol; PI3K: Phosphoinositide 3-kinase; RoA: Route of administration; ROS:Reactive oxygen species; SOD: Superoxide dismutase; S-ODN: Antisense phosphorothioate oligodeoxynucleotide; TBARS: Thiobarbituric acid reactive substances; TG: Triglycerides; TGFβ: Transforming growth factor beta; TIMPs:Tissue inhibitors of metalloproteinases; TNFα: Tumor necrosis factor alpha; ZO1: Zonal occludin 1.
Liposome-mediated drug delivery in ALD
Liposomes are one of the most common targeted drug delivery platforms, and indeed, about a third of the studies reviewed here used liposomal drug delivery in some form. Liposomes are vesicles composed of a phospholipid bilayer consisting of one (unilamellar) or more (multilamellar) concentric spherical layers enclosing an aqueous center (Figure 2A, middle panel)[23]. The presence of both aqueous and lipid compartments allows encapsulation or attachment of large quantities of both hydrophilic and lipophilic drugs, respectively (even simultaneously). Liposomes can be modified in many ways to alter their biodistributionin vivo, for example by modifying the lipid composition (saturatedvsunsaturated,positively chargedvsnegatively charged), controlling size, attaching molecules such as PEG to improve stability, or adding proteins, antibodies, peptides, or carbohydrates to facilitate targeting of a specific cell type. The use of naturally occurring phospholipids gives liposomes the advantage of typically being non-immunogenic and non-pharmacologically active when administered alone. A major challenge in using liposomes to deliver drugs to the liver is opsonization and clearance by KCs and LSECs, as well as by RES components in the liver and other organs including the spleen, kidney, lung, bone marrow, and lymph nodes, although the liver is the primary site of liposome retention[23]. Attaching PEG to the liposome surface is an effective way to improve pharmacokinetics and avoid RES clearance, as PEG prevents attachment of opsonizing molecules and subsequent recognition by macrophages[19].Controlling liposome size and surface charge can also avoid opsonization, as smaller (approximately 200 nm), more neutral liposomes do not as readily bind plasma proteins as larger, more highly charged liposomes.

Figure 4 Schematic representation of literature search strategy. Initial search terms included “alcohol liver disease” combined with the boxed terms.Eight hundred and forty-six unique results were generated, screened by title and abstract, and excluded based on relevance to the scope of the review. Sixteen studies were included in the review, broken down into four categories based on drug delivery platform. ALD: Alcohol-associated liver disease; AAV: Adeno-associated virus.
An early study by Ponnappaet al[24] used pH-sensitive liposomes consisting of phosphatidylethanolamine, cholesterol hemisuccinate, and cholesterol to encapsulate an antisense oligonucleotide againstTnfmRNA (termed S-ODN) for delivery to the liver in a passive targeting approach. TNF-α is a proinflammatory cytokine elevated in ALD which, at high concentrations, sensitizes hepatocytes to cell death signals[25]. Liver macrophages and monocytes are a large source of liver TNF-α production[26].Given the ability of liposomes to passively target liver macrophages, liposomes were therefore a natural choice of platform for the authors to employ in order to increase delivery of S-ODN to KCs. Intravenous administration of liposomal S-ODNs in a rat chronic ALD model decreased liverTnfmRNA expression as expected, and subsequently prevented liver injury as demonstrated by plasma ALT[24]. The concentration of S-ODN in KCs was confirmed as being 20-fold higher compared to hepatocytes. In that study,liver-targeted delivery of the therapeutic was necessary to prevent side effects, specifically, to avoid the inhibition of blood coagulation associated with systemic administration (a process already perturbed in liver diseases[27]). A study by Rodriguezet al[28] also used a liposomal delivery system to avoid the systemic side effects of the hepato-protective drug rolipram, a phosphodiesterase 4 inhibitor. Previous studies demonstrated the beneficial effects of rolipram for ALD and other liver diseases[29]. However,in humans, rolipram causes significant central nervous system and gastrointestinal side effects(headache, vomiting,etc.). To this end, Rodriguezet al[28] used fusogenic liposomes composed of 1,2-Dioleoyl-sn-glycerol-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphate to passively deliver rolipram to the liver. Fusogenic liposomes differ from conventional liposomes because they avoid endocytosis and lysosomal degradation and instead fuse with the target cell membrane to release the drug cargo into the cytoplasm (for hydrophilic drugs) or membrane (for lipophilic drugs)[30]. Since phosphodiesterase 4 is expressed in the cytoplasm and plasma membrane of HSCs, among other liver cell types[31], the fusogenic liposome platform was an obvious choice for rolipram delivery. Indeed, in an acute-on-chronic mouse model of ALD, rolipram-loaded liposomes reduced liver damage (as determined by plasma ALT/AST activity), steatosis, oxidative stress, and endoplasmic reticulum (ER)stress similar to unencapsulated rolipram. However, encapsulated rolipram prevented liver cell death to a greater degree than un-encapsulated rolipram.
In 2016, Zhaoet al[32] employed a liposome approach to deliver puerarin to the liver. Unlike the previous two studies, liposomal encapsulation in this study was used to improve pharmacokinetics,because puerarin, a plant-derived isoflavin, is rapidly cleared from the blood by the kidneys (with a half-life of less than one hour[33]). Liposomal encapsulation of this hydrophilic drug was achieved with liposomes composed of phosphatidylcholine, cholesterol, and propylene glycol. The authors demonstrated improved pharmacokinetics when administering puerarin liposomes to mice compared to non-encapsulated puerarin. Specifically, plasma area under the curve and half-life improved by 2.37-and 4.16-fold, respectively, and puerarin was detected most highly in the liver compared to other organs in both preparations. Previous studies supported puerarin as a beneficial molecule in a rat ALD model[34], but liposomal encapsulation improved efficacy further with respect to liver injury (decreased plasma ALT and AST levels) and, to a lesser degree, steatosis. In 2019, Wuet al[35] similarly employed a liposomal encapsulation technique to improve the pharmacokinetics of a naturally produced antiinflammatory carotenoid, astaxanthin. Liposomal astaxanthin was administered to mice either orally or by intraperitoneal injection in an intragastric ethanol feeding model of ALD. Oral and intraperitoneal liposomal astaxanthin ameliorated alcohol-induced liver injury and histological signs of fibrosis.Whereas biodistribution of astaxanthin liposomes was not directly characterized in this study, the physical properties of the drug (low bioavailability, poor water solubility) suggest that liposomal encapsulation was necessary for efficacy. Silymarin is another excellent example of a beneficial compound with poor pharmacokinetics which can be improved by incorporation into liposomes. Kumaret al[36] showed that encapsulation of this hepato-protective flavonolignan in phosphatidylcholine and cholesterol liposomes (either un-modified or PEGylated) improved pharmacokinetics and efficacy.Liposomal encapsulation improved the maximum plasma concentration and plasma area under the curve, while also increasing the solubility of the drug.In vitro, silymarin liposomes protected Chang Liver (HeLa) cells against ethanol-induced cell death.In vivo, in a rat chronic model of ALD, both unmodified and PEGylated silymarin liposomes ameliorated alcohol-induced liver injury while retaining the anti-inflammatory and antioxidant properties of silymarin. Recently, Yuet al[37] encapsulated Saikosaponin D, an anti-inflammatory/anti-oxidant plant-derived compound, in liposomes and demonstrated improved pharmacokinetics and efficacy in a mouse model of ALD compared to the nonencapsulated compound.
Lastly, Jainet al[38] employed a liposomal encapsulation approach for a plant-derived molecule,mangiferin. Like silymarin, mangiferin is a natural antioxidant with demonstrated benefits in the treatment of ALD and other diseases[39,40], but is not efficacious when used alone due to low bioavailability and metabolism by gut bacteria, as demonstrated by Jainet al[38]. To this end, the authors used a so-called ‘herbosome’ encapsulation strategy for mangiferin to improve the bioavailability of this compound. Herbosomes are defined as plant-derived compounds encapsulated in phospholipid particles, which in this study consisted of phosphatidylcholine and cholesterol. In a chronic rat model of ALD, unencapsulated mangiferin was able to significantly decrease liver injury, and mangiferin-loaded herbosomes further decreased the liver injury. Mechanistically, the authors attributed this protection to the antioxidant effects of mangiferin, as demonstrated by rescued SOD, catalase, and GSH levels and decreased liver MDA.
One of these blocks had been placed by loving hands on a child s grave, and one of the women who had come out of the church walked up to it; she stood there, her eyes resting on the weather-beaten memorial, and a few moments afterwards her husband joined her
These studies support liposomal encapsulation as an effective approach not only for targeting drugs to the liver to avoid systemic side effects, but also for increasing the bioavailability of various compounds. The studies cited herein accomplished these goals by using liposomes composed of various glycerophospholipids including phosphatidylethanolamine, phosphatidyl choline, phosphatidic acid,and lecithin. Selection of certain lipids over others influences membrane fluidity/rigidity, which indirectly alters the permeability of the liposomal bilayer[41]. Certain phospholipids can also be chosen over others to impart fusogenic character, wherein liposomal cargoes can be targeted to the cell cytoplasm by fusing with the plasma membrane while avoiding endocytic degradation[30]. Rodriguezet al[28] employed this approach by using liposomes composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate to target cytosolic phosphodiesterase.Further, most other groups incorporated cholesterol into their formulation, which can alter the release of the drug cargo and prevent unwanted ‘leakage’, thereby contributing to the overall stability of the nanoparticle[42].
Exosomes in ALD
Exosomes are another type of lipid-based liver-targeting nanoparticle which have been evaluated preclinically as potential therapeutics for ALD as well as potential biomarkers of disease progression in AH[43] (Figure 2A, right panel). Exosomes can be derived from bacteria or food, and are often small(approximately 30-150 nm in diameter) compared to synthetic liposomes (150 nm and larger in the studies cited here)[44]. Because they are products of the host cell membrane which are excreted by exocytosis, they are composed of phospholipids and cholesterol. While originally thought to be used by cells for waste removal, more recent evidence has supported a role in cell signaling, antigen presentation, tissue repair and regeneration, among other processes[44]. Unlike liposomes, exosomes contain numerous surface proteins (e.g., CD63 in eukaryotic exosomes[45]) and internal cargo molecules including lipids, proteins, and nucleic acids. Despite the presence of existing cargo, additional molecules including drugs can be added to exosomes after isolation. The surface proteins present on exosomes mediate their cellular uptake, which has been shown to occur mostly in the liver and spleen, but also to some degree in the kidney, lung, and gastrointestinal tract, although pharmacokinetics depend on the source of the exosomes[46]. Liver macrophages are the cell type thought to be most responsible for exosome uptake through recognition of their charge by scavenging receptors, or recognition of surface signals such as sialic acid or phosphatidyl serine[47,48]. Thus, clearance by macrophages is again a drawback when trying to administer drugs to the liver parenchyma. Another significant consideration is standardization of isolation or purification protocols. Some techniques, for example, fail to completely exclude extraneous types of extracellular vesicles, leading to an impure drug product preparation[49].Lastly, there are many unanswered questions related to how the choice of cell type from which to isolate exosomes impacts immunogenicity and efficacy.
A study from Guet al[50] aimed to use exosomes derived from the beneficial bacteriaLactobacillus rhamnosus GG(LGG) to treat ALD. LGG has previously been demonstrated to be beneficial for ALD as a probiotic supplement which prevents gut permeability, thereby ameliorating liver injury[51,52]. The benefits of LGG probiotic supplementation are mediated, in part, by molecules secreted by LGG, as evidenced by the protective effects of LGG cell culture supernatant[53,54]. These soluble mediators are thought to be released from bacteria in exosomes. Guet al[50] showed that orally administered LGGderived exosomes (termed LDNPs) ameliorated experimental ALD in an acute-on-chronic mouse model. In contrast to previous studies aiming to deliver drugs to the liver, LDNPs in this study were designed to ameliorate liver injuryviathe gut-liver axis, by targeting intestinal cells. Fluorescent labeled LDNPs were detectable in the intestine to a much larger extent than in the liver. Mice that received LDNPs were protected from the ethanol-associated reduction in intestinal tight junction protein expression and had boosted expression of intestinal anti-microbial peptides (e.g.,Reg3b,Reg3g) and IL-22. As a result, circulating endotoxin levels were decreased in LDNP-treated mice. Consequently, liver injury, steatosis, and inflammation were attenuated, confirming the critical importance of intestinal barrier defense in preventing ALD pathogenesis. Mechanistically, Guet al[50] showed that the beneficial effects of LDNPs were mediated by the aryl hydrocarbon receptor (AhR), suggesting that the cargo molecules responsible for the benefits of LDNPs are likely AhR ligands.
Foods are another excellent source of exosomes with beneficial endogenous cargo molecules which target the intestinal epithelium or translocate to the bloodstream to target various organs including the liver[55]. Fluorescently labelled milk-derived exosomes, for example, have been shown to localize in the liver after oral administration to mice[56]. Food-derived exosomes from ginger, grapefruit, grape, garlic,ginseng, lemon, and others have been shown to be efficacious in the treatment of numerous diseases by nature of their antioxidant, anti-tumor, or anti-inflammatory cargo[55]. To investigate the efficacy of food-derived exosomes in ALD, Zhuanget al[57] used exosomes derived from ginger, a food which has been demonstrated to protect against liver injury of multiple etiologies, including alcohol,viaantioxidant compounds called gingerols[58]. In an acute-on-chronic mouse model of ALD, daily oral ginger-derived exosome delivery decreased liver injury and steatosis. The antioxidant effects of the exosomes were also demonstrated, with increased expression of antioxidant genes in the liver through activation of NRF2. The authors also analyzed the distribution of the exosomes by fluorescent labeling,showing that the liver was the primary site of accumulation, with no detectable signal in lung, spleen, or other organs. Further, co-localization with albumin-positive cells by immunofluorescence showed that the ginger-derived exosomes primarily associated with hepatocytes, indicating cell-specificity.Collectively, these studies show the utility of exosomes as ‘pre-packaged’ lipid vesicles which can deliver beneficial cargo molecules from various sources to the liver for the treatment of ALD.
PNP-mediated drug delivery in ALD
PNPs are a class of non-lipid-based nanoparticles composed of natural or synthetic polymers that are gaining popularity in numerous applications, including medicinal and non-medicinal (material science,electronics, ecology,etc., Figure 2B, left panel)[59]. PNPs are classified as either nanospheres (composed entirely of polymer matrix) or nanocapsules (a polymer shell with a water or oil center) with an approximate size of 100-250 nm, which can be controlled during synthesis. Poly(lactic-co-glycolic acid)(PLGA) and vinyl monomer-based polymers are commonly used in PNP synthesis (e.g., polystyrene,polyalkyl acrylates), although many other polymers can be used including polyesters, polyurethanes,polysaccharides, polypeptides, and biopolymers (e.g., lignin)[60]. Polymer choice can be adjusted to control stability, particle size, andin vivodrug release. As with liposomes, surface modifications can also be made to PNPs to alter their pharmacokinetic profile and biodistribution, such as active targeting moieties or hydrophilic molecules that prevent opsonization (e.g., PEG). Surface modifications can also change the intrinsic negative charge of most PNPs to neutral or positive. PEGylation, for example, shifts the charge to neutral, whereas conjugation of other molecules such as chitosan imparts a positive charge[61,62]. After reaching target cells, PNPs are up taken by pinocytosis or clathrin-mediated endocytosis but can escape lysosomal degradation and enter the cell cytoplasm within 10 min[63]. Other benefits of PNPs include low immunogenicity, low toxicity, and large surface area. As with liposomes, one drawback of PNPs is their susceptibility to opsonization in plasma and rapid clearance by the liver and spleen RES.
Several studies have applied PNPs to the treatment of ALD by attaching various cargo molecules. A study by Naget al[64] used PLGA PNPs to deliver tannic acid and vitamin E to the liver in a chronic mouse model of ALD. These two naturally occurring molecules have previously been established to be beneficial for the treatment of ALD through anti-inflammatory and antioxidant mechanisms[65]. PNP formulation is necessary to ensure extended release of these molecules due to intestinal modification,poor absorption, rapid metabolism, and short half-life[66,67]. Naget al[64] demonstrated that tannic acid/vitamin E PLGA PNPs ameliorated ALD as evaluated by multiple endpoints including reduced liver injury, steatosis, fibrosis, inflammation, oxidative stress, and liver cell apoptosis, as well as increased hepatocyte viability. Importantly,in vitropharmacokinetic analysis showed that the PNPs slowed the movement of tannic acid and vitamin E across a semi-permeable membrane compared to free tannic acid and vitamin E, indicating that this formulation may improve the retention time of these compounds in the liver.
Another study targeting oxidative stress in ALD was conducted by Natarajanet al[68], who employed a PNP approach to deliver the enzyme superoxide dismutase (SOD) to the liver. Oxidative stress is a key mechanism in ALD pathogenesis[69]. A previous study in rats demonstrated that increasing hepatic SOD expression (viagene therapy) alleviated ALD by scavenging superoxide[70]; however, PNPmediated SOD delivery is a more favorable translational therapy due to clinical issues surrounding the use of gene therapy (hepatotoxicity and generation of anti-adenovirus antibodies, for example)[71].Further, previous studies suggest that administration of unencapsulated recombinant SOD does not produce effects that are as long-lasting as those by encapsulated SOD[72]. After establishing successful delivery of functional SODin vitroin E-47 hepatocytes and protection against ethanol and linoleic acidinduced oxidative stress, the authors administered SOD PNPs to mice by intraperitoneal injection in a chronic model of ALD. Compared to ethanol-treated mice, mice which received ethanol and SOD PNPs had decreased liver steatosis and inflammation as quantified by hematoxylin-eosin staining and decreased liver cytokine expression, respectively. Interestingly, the authors could not detect an increase in SOD in SOD PNP-treated mice, although the authors speculate that the time course of the study may not allow proper detection of elevated SOD levels. In a follow up study by the same research group,Gopalet al[73] again assessed the efficacy of intraperitoneal administered SOD PNPs in ALD, although in a modified model where mice are fed a high fat diet prior to the beginning of the ethanol feeding paradigm. Unlike the previous study, here the authors were able to show evidence of increased SOD expression and activity in the livers of mice administered SOD PNPs. Ethanol significantly induced liver injury in control mice, but not in mice administered SOD PNPs, as evidenced by plasma ALT levels.Again, ethanol-induced hepatic steatosis and inflammation were attenuated, corroborating the beneficial effects and mechanisms of protection of liver-specific SOD delivery.
Apart from proteins, another group of novel cargo molecules which can be delivered by PNPs are anti-micro RNAs (anti-MIRs), which are designed to inhibit endogenous MIRs, such as MIR-155, which has been previously shown to play a pathogenic role in ALD[74]. Zhanget al[75] aimed to not only block the effects of MIR-155, but also to deliver CXCR4 antagonists (collectively termed polycationic CXCR4 antagonists, or PCX, by the authors), which block alcohol-induced liver fibrosisviainhibition of HSC activation. Thus, the group administered synthetic cholesterol-modified polyethyleneimine nanoparticlesviai.v. injection to mice to target HSCs and KCs in a model of alcohol + CCL4-induced fibrosis. Indeed, compared to nanoparticles harboring a MIR negative control, the anti-MIR-155/PCXloaded PNPs significantly reduced liver injury, fibrosis, and inflammation when administered in a treatment paradigm. This study supports the idea that numerous therapeutic cargos are compatible with the PNP platform, even when combined in a dual approach.
In contrast to the synthetic PNPs used in the studies mentioned above, a study by Wanget al[76].used PNPs synthesized from a naturally occurring polysaccharide isolated fromAngelica sinensisroot [Angelica sinensispolysaccharide (ASP)]. ASP was combined with cholesterol hemisuccinate to prepare self-assembling ASP-cholesterol hemisuccinate PNPs (termed ACNPs), which were loaded with curcumin, a plant-derived compound with antioxidant effects which has previously shown beneficial effects in ALD[77,78]. The authors used PNPs to improve the delivery of curcumin, which is not readily water-soluble and has low bioavailability due to rapid metabolism[79]. In an intragastric feeding mouse model of ALD, the authors demonstrated that curcumin-loaded ACNPs decreased liver oxidative stress,and consequently, liver injury. Mechanistically, curcumin ACNPs increased NRF2 protein, consistent with other studies implicating NRF2 signaling for the beneficial effects of curcumin[77,80]. These studies show that PNPs, in addition to liposomes, are an effective choice of delivery platform to target drugs to the liver, and importantly, improve the bioavailability of compounds such as tannic acid,vitamin E, and curcumin.
Bacteria and adeno-associated virus-mediated liver-specific delivery in ALD
Certain bacteria have long been considered for their therapeutic potential either as whole organisms(probiotics), colonies of many bacterial species (i.e., fecal transplant), or bacterial products[81]. More recently, genetically engineered bacteria have been developed to facilitate delivery of drugs, proteins,enzymes, and genes for the treatment of numerous pathologies[81] (Figure 2C, left panel). Bacteria as drug delivery systems are beneficial in several ways, including that they can provide their own propulsion and taxisviaflagella or pili in response to external stimuli (e.g., phototaxis, chemotaxis,thermotaxis,etc.), they can be designed to seek a certain molecule (i.e., active targeting), they can produce a desired drug ‘on-site’ by metabolism, and they can even be designed to transfect host cells.These benefits, and numerous others (described in great detail in[81]), come at the cost of potential host immune response. Use of non-pathogenic bacterial strains, commensal bacteria, or genetic modification can decrease immunogenicity, but there is still considerable risk of septic shock which can result in mortality when targeting sterile body compartments (e.g., blood, abdominal cavity,etc.)[82]. Hendrikxet al[83], for example, used genetically engineeredLactobacillus reuteri(L. reuteri), a commensal gut microbe, as a means of increasing intestinal IL-22 for the treatment of ALD in an acute-on-chronic mouse model. This approach aimed to ameliorate alcohol-associated changes in both the intestine and the liver. The gut and liver are connectedviathe so-called gut-liver axis, where alcohol-induced gut permeability allows pro-inflammatory bacteria and bacterial products (e.g., endotoxins) to enter the hepatic portal system and exacerbate liver injury[84]. IL-22 is a cytokine which contributes to gut barrier defense and homeostasis[85], which the authors demonstrated to be decreased in the intestines of ethanol-fed mice. Mice which were enterally provided IL-22-expressingL. reuterithroughout the feeding protocol had increased expression of the gut anti-microbial peptide,Reg3g, decreased translocation of bacteria to the liver, and consequently, decreased liver injury, steatosis, and inflammation. Increased intestinal IL-22 was confirmed, but there was no increase in plasma IL-22, indicating that a localized increase in gut IL-22 was sufficient to restore gut barrier health and ameliorate liver injury. This engineered bacteria approach may be more clinically useful than simply administering recombinant IL-22 protein systemically, as systemic administration is associated with increased risk of tumor development in chronic liver disease patients[86-88]. Indeed, bacteria serve as a unique drug delivery system with several key advantages, especially given that liver diseases such as ALD can be targeted indirectlyviathe gut-liver axis.
AAVs (adeno-associated virus) vectors are another biological system with the capability to target specific organs (Figure 2C, right). There exist multiple AAV serotypes with differing capsid proteins (13 in total), which confer serotype-specific functional features, including tropism for different organs[89].AAV serotype 8 (AAV8), for example, exhibits high liver tropism, since the capsid proteins expressed in this serotype interact with the laminin receptor, which is highly expressed in the liver (for this reason,we have defined AAV8 as ‘actively’ targeting the liver in Table 2)[90]. This natural ability to target the liver comes at the cost of immunogenicity, liver toxicity, and the production of neutralizing antibodies by the host, several key hurdles for clinical AAV8-based therapy[89]. In contrast to the previous nanoparticle- and bacteria-based delivery systems discussed in this review, AAV vectors are used as a gene delivery vehicle, rather than a carrier of natural or synthetic drugs, based on their ability to transfect host cells[91]. This review will discuss one study using AAV8 as a delivery mechanism for a microRNA; for more information regarding gene therapy for the treatment of liver disease the reader is encouraged to read Kattenhornet al[92]. Satishchandranet al[93] employed an AAV8 vector to rescue the ethanol-associated loss of microRNA 122 (MIR-122), which was demonstrated in both human ALD patients and mice in a 5-week chronic model of ALD. Previous work showed that loss of liverMir122alone led to hepatic steatosis with spontaneous development of liver fibrosis and even HCC[94],suggesting a beneficial or homeostatic role of this microRNA in the liver. The authors used the AAV8 serotype to transfect hepatocytes withpri-MiR122or a scrambled control vector. Compared to controls,mice receiving AAV8-MIR122 had increased mature liver MIR-122 and, importantly, decreased alcoholinduced liver injury, steatosis, inflammation, and fibrosis. The AAV8 vector was shown to specifically target hepatocytes (as liver mononuclear cells had no increase in MIR122), suggesting this platform may be effective in targeting genes, including microRNAs, to not only the liver, but specifically to hepatocytes.
GAPS IN KNOWLEDGE
Research efforts to apply targeted drug delivery systems for the treatment of ALD are growing, but there are still considerable gaps in knowledge and several barriers to address. First is the lack of use of active targeting strategies, where addition of a ligand to a liposome or nanoparticle targets a drug to a particular liver cell type. Targeting a drug to a particular cell type (e.g., targeting an antioxidant to hepatocytes) may increase efficacy, or produce the same beneficial effect with a lower total dose, thereby reducing the possibility of off-target effects. In additional to hepatocytes, other cell types contribute to ALD, including HSCs and both resident (Kupffer cells) and infiltrating macrophages, thus presenting opportunities to target these non-parenchymal liver cells. In this way, such treatments could target various stages of ALD such as hepatic fibrosis, which is largely driven by these cell types in their activated states (i.e., activated HSCs or M1-polarized KCs)[95]. Fifteen of the 16 studies used a passive targeting approach, where the physical properties of the particle (i.e., size and charge) were controlled in such a way that the particles would passively accumulate in the liver. The Satishchandranet al[93] study using AAV8 is one example of biological active targeting, where the AAV8 capsid binds to a particular receptor in the liver. Future research should consider ligand conjugation and active targeting of liposomes and PNPs to improve their drug formulations.
Next, with respect to the paradigm in which drug therapies were administered, in the 16 studies reviewed here, only about half used a so-called ‘treatment paradigm’, where the drug was given after establishment of liver injury (i.e., half-way through the model or later). The remaining half administered their therapeutics in a ‘prevention paradigm’, where the drug was given at the start (or even prior to the start) of the alcohol feeding model. Studying the efficacy of a drug in a prevention paradigm certainly provides useful insight into whether the drug has any beneficial effect in ALD. However, this paradigm has limited clinical relevance, since most patients with mild to moderate ALD are asymptomatic, and they would not receive a diagnosis nor treatment until after injury has developed. In the case of studies establishing benefits of a liver-targeted drug in ALD in a prevention paradigm, additional studies should be carried out to determine whether administration of that drug formulation later in the feeding model is still effective. Another issue related to the models used in these studies is the lack of knowledge of the efficacy of these therapies in advanced ALD stages such as fibrosis/cirrhosis. Only one study discussed here (Zhanget al[75]), employed a model which is known to produce liver fibrosis,in this case by use of a ‘second hit’ of carbon tetrachloride superimposed on chronic EtOH feeding.While the authors did note reductions in fibrosis as measured by immunohistochemistry, this is only one study. Most of the studies discussed herein employed chronic, acute-on-chronic, or multiple-binge models which typically produce mild ALD characterized by hepatic steatosis, low-level inflammation,and mild liver injury with elevated ALT but no fibrosis[96,97]. Future studies should investigate the efficacy of nanoformulated drugs in experimental models of more advanced ALD which mimic alcoholassociated cirrhosis or severe AH, especially as better models are developed.
Another consideration with significant clinical implications is the route of administration. The studies reviewed here applied oral (gavage) or injection routes of delivery. Clearly, oral delivery is most attractive from a patient compliance perspective due to ease of self-administration and the absence of potential adverse effects from injections (injection site pain, inflammation, and infection). In general,nanoparticle systems tend to improve the pharmacokinetics of a drug to enable oral delivery in cases where this route would be otherwise unfeasible due to enzymatic digestion or poor absorption[98].Indeed, many of the studies discussed here employed these platforms with this goal in mind, particularly for poorly soluble plant-derived compounds. However, compared to oral delivery, injection allows the highest level of control over the rate of drug delivery and can bypass any issues associated with first-pass metabolism or poor gastrointestinal absorption, resulting in a bioavailability of 100% and a rapid onset of action[99]. Direct injection of the drug solution into circulation does, however, pose a higher risk of adverse reactions and requires a healthcare professional to administer the treatment. This may be most acceptable in cases where drugs are developed for advanced ALD stages such as AH,where patients are already hospitalized. Regardless, authors should justify their chosen route of administration in the context of their future translational goals.
Additionally, in pre-clinical ALD research, it is important to consider sex differences, since men and women consume and metabolize alcohol differently, have different risk factors contributing to ALD,and ultimately, have different susceptibility to developing the disease[100]. Even in mice, there are sex differences in susceptibility to ALD when controlling for alcohol intake, diet, and other factors[101].Further, evidence suggests biodistribution of nanoparticles may also differ by sex[102], providing an additional rationale for studying nanoparticle systems in ALD in both sexes. Despite these differences,many of the studies reviewed here (14 of 16) used either only male mice or only female mice, and the remaining two which used both sexes did not report the results for each sex separately.
Lastly, keeping in mind the goal of translating effective therapies to humans for the treatment of ALD, there is a lack of knowledge regarding the efficacy of liver-targeted therapies in humans for this disease. Critically, however, nanoparticle platforms have been used for many years for the treatment of other diseases. For example, liposomes have been used in numerous drug formulations for the treatment of various cancers, fungal and viral infection, pain, and other diseases since 1995 with excellent safety and efficacy[103]. Liposomes are also increasing in popularity as an excellent vaccine delivery system with several benefits over conventional vaccines (e.g., liposomes are used in the Moderna and Pfizer/BioNTech COVID-19 mRNA vaccines)[104]. Although less common than liposomes, PNPs have also undergone clinical evaluation for the treatment of head, neck, lung, and breast cancers[105]. Other platforms not discussed in this review, such as N-acetyl-galactosamine(GalNac) conjugate (commonly used to delivery nucleic acids to hepatocytes by binding the asialoglycoprotein receptor[106]), have also been shown to have favorable safety profiles in clinical trials[107].Clearly, drug delivery platforms with the capability to deliver drugs to the liver have undergone significant clinical evaluation, although not for the treatment of ALD. Future work should build on the growing pre-clinical data supporting the efficacy of particle therapeutics in ALD and the existing clinical data showing the safety of these systems in humans to move these nanomedicines to the clinic.
CONCLUSION
The research efforts reviewed here employed liver-targeted (or intestine-targeted) drug delivery platforms to improve their drug formulations and more effectively develop pharmacological interventions for ALD (summarized in Figure 5). These platforms, including liposomes, PNPs,exosomes, bacteria, and AAV vectors are aimed at improving a drug’s pharmacokinetics, efficacy, and safety by reducing off target effects associated with systemic delivery and increasing the concentration of the drug locally in the liver. The authors of these studies used nanomedicine platforms to deliver phosphodiesterase inhibitors, naturally occurring antioxidants, oligonucleotides, miRNAs, enzymes,and anti-inflammatory cytokines in various rodent models of ALD, showing promising results which will move the pace of drug development for this disease forward toward clinical translation. Future studies should continue to apply and characterize targeted delivery platforms, as well as consider active targeting approaches, drug administration paradigms, and sex-specific differences in the pursuit of supporting future clinical trials in this field.

Figure 5 Summary of studies applying nanoparticle platforms in alcohol-associated liver disease. A graphical summary of nanoparticle platforms which to date have been applied for the treatment or prevention of alcohol-associated liver disease in rodent models. Arrows represent organ targets of each platform.Example cargoes used in research articles discussed in this review are listed next to each platform. Current limitations of the field are described on the lower right.AAV8: Adeno-associated virus serotype 8; HSCs: Hepatic stellate cells; IL-22: Interleukin 22; KCs: Kupffer cells; miRNA: Micro-RNA; PCX: Polycationic CXCR4 antagonists; S-ODN: Antisense phosphorothioate oligodeoxynucleotide.
ACKNOWLEDGEMENTS
We apologize to and acknowledge any authors whose studies were not identified by our literature search strategy. We also acknowledge Marion McClain for manuscript editing support.
FOOTNOTES
Author contributions:Warner JB and Kirpich IA designed and outlined the review; Warner JB wrote the contents of the review; Warner JB and Guenthner SC designed the figures; Guenthner SC, Hardesty JE, McClain CJ, Warner DR,and Kirpich IA edited the manuscript; all authors have read and approve the final manuscript.
Supported byNational Institutes of Health, No. R01AA028905-01A1 (to Kirpich IA), No. 1F31AA028423-01A1 (to Warner JB), No. F32AA027950-01A1 (to Hardesty JE) and No. U01AA026934 (to McClain CJ); Jewish Heritage Fund for Excellence Research Enhancement Grant Program at the University of Louisville, as well as an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health, No. P20GM113226 (to McClain CJ); and National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health, No. P50AA024337 (to McClain CJ).
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORCID number:Jeffrey Barr Warner 0000-0003-2022-7854; Steven Corrigan Guenthner 0000-0003-3561-9607; Josiah Everett Hardesty 0000-0003-1955-3046; Craig James McClain 0000-0002-7219-8939; Dennis Ray Warner 0000-0002-5303-6870; Irina Andreyevna Kirpich 0000-0002-9545-6451.
S-Editor:Gao CC
L-Editor:A
P-Editor:Gao CC
 World Journal of Gastroenterology2022年36期
World Journal of Gastroenterology2022年36期
- World Journal of Gastroenterology的其它文章
- Nonalcoholic steatohepatitis and hepatocellular carcinoma: Beyond the boundaries of the liver
- Impact of sarcopenia on tumor response and survival outcomes in patients with hepatocellular carcinoma treated by trans-arterial (chemo)-embolization
- Early extrahepatic recurrence as a pivotal factor for survival after hepatocellular carcinoma resection: A 15-year observational study
- SARS-CoV-2 and the pancreas: What do we know about acute pancreatitis in COVID-19 positive patients?
- Esophageal magnetic compression anastomosis in dogs
- P2X7 receptor as the regulator of T-cell function in intestinal barrier disruption