
Chromosome-level genome assembly of Tibetan macaque (Macaca thibetana) and species-specific structural variations
2022-10-17 03:27Ru-SongZhang,ChuangZhou,Xian-LinJin等
Zoological Research 2022年5期
DEAR EDITOR,
The Tibetan macaque (Macaca thibetana) is an endemic species in China belonging to thesinicagroup in the genusMacaca. Within the genus, it is the largest in body size and has a wide geographic distribution, with a population size second only to rhesus macaques (M. mulatta). Here, using PacBio long-read sequencing and Hi-C technology, we assembled the first chromosome-level genome of the Tibetan macaque, with a size of 2.82 Gb, contig N50 of 48.75 Mb, and scaffold N50 of 150.62 Mb, anchored to 22 chromosomes.This assembled genome is of higher quality than previously published chromosome-level genomes of rhesus and cynomolgus macaques. In total, 22 485 protein-coding genes and 1.33 Gb of repeat sequences were annotated in the Tibetan macaque genome. Phylogenetic analysis indicated that the Tibetan macaque was closely related to the stumptailed macaque and diverged from a common ancestor 3.59 million years ago (Ma). A total of 454 positively selected genes(PSGs) were identified, which were enriched in pathways related to protein binding, anatomical structure development,and developmental processes. Among them, four genes associated with tail development and eight genes associated with body size were under positive selection, which may contribute to their short tail and large body size. Structural variation (SV) analysis between the Tibetan macaque and other macaque species identified 6 778 Tibetan macaquespecific SVs (TMSSVs). Among these SVs, three deletions and four insertions in six genes may be associated with tail development and body size. This high-quality Tibetan macaque genome should benefit further biological and evolutionary studies on primates.
According to Roos et al. (2014), species in the genusMacacacan be divided into seven species groups: i.e.,sylvanus,silenus,sulawesi,sinica,arctoides,fascicularis, andmulatta. Among them,de novoassembled genomes of representative species from thesilenus(pig-tailed macaques),arctoides(stump-tailed macaques),fascicularis(cynomolgus macaques), andmulattagroups (rhesus macaques) have been reported. At present, chromosome-level genome assemblies based on long-read sequencing and Hi-C technologies are only available for rhesus (GCA_003339765)and cynomolgus macaques (M.fascicularis)(GCA_011100615), while scaffold-level assemblies based on short-read sequencing data are available for Japanese (M.fuscata) (GCA_003118495), pig-tailed (M. nemestrina)(GCA_000956065), and stump-tailed macaques (M. arctoides)(GCA_021188215). Genomes for thesinicagroup species are not currently available. We previously reported a ~37X coverage genome of the Tibetan macaque based on shortread sequencing technology to identify species-specific variants (Fan et al., 2014). Although short-read sequencing assemblies provide useful information regarding genomic variation, they cannot provide large-scale structural information and variation due to technology limitations. A highquality chromosome-level Tibetan macaque reference genome is still needed to better understand adaptive evolution and perform related research. Thus, we constructed a highquality Tibetan macaque genome assembly using PacBio long-read sequencing and Hi-C technology. We annotated the newly assembled genome, performed comparative genomic analysis with other macaque species, and identified TMSSVs.This study provides a valuable resource for future investigations on genomic diversity and adaptive evolution in primates.
The animal used for sequencing was an adult Tibetan macaque (male, 9 years old) originally from Mabian Dafengding National Nature Reserve and now housed in Sichuan Hengshu Biotechnology Co., Ltd. (Yibin, China).Sample collection followed the Regulations for the Implementation of the Protection of Terrestrial Wildlife of China (State Council Decree [1992] No. 13) and was approved by the Ethics Committee of the College of Life Sciences, Sichuan University, China (Permit No.20200327009). Detailed descriptions of sequencing,de novoassembly, genome annotation, and comparative genomic analysis are provided in the Supplementary Materials and Methods.
For short-read DNA sequencing, clean short reads (120 Gb)were obtained by removing low-quality and redundant reads.We then estimated Tibetan macaque genome properties,heterozygosity, and repeat content using GCE (v1.0.2).Genome size was estimated to be 3.08 Gb, with a heterozygosity of 0.28% and repeat content of 52.31%.However, genome size may be overestimated, as estimated size is always larger than the actual value (Liu et al., 2013). As shown in the 17-mer depth distribution curve, the Tibetan macaque genome had a high repeat content (Figure 1A).
For long-read sequencing, PacBio Sequel II generated a total of 230.3 Gb of data (~86-fold coverage), with an average length and a N50 length of subreads of 22 108 bp and 39 020 bp, respectively. The contig set generated by Flye was selected for post-scaffolding using Hi-C as it outperformed MECAT2 and wtdbg2 in terms of BUSCO (Benchmarking Universal Single-Copy Orthologs) completeness, CEGMA(Core Eukaryotic Genes Mapping Approach) completeness,and contig continuity (Supplementary Tables S1-S3). Flyebased assembly and polishing with Racon and Pilon yielded an initial assembly of 2.82 Gb with a contig N50 of 48.75 Mb and longest contig of 103.89 Mb (Supplementary Table S4).
A total of 398.5 Gb (~148-fold coverage) of Hi-C reads were obtained to assist in anchoring contigs to chromosomes.Finally, 291 contigs were successfully clustered, ordered, and oriented on 22 chromosomes (Figure 1B), with a total length of unplaced contigs of only 11.88 Mb (0.42%). The scaffold N50 of the Tibetan macaque genome was 150.62 Mb, and the longest scaffold was 220.36 Mb. The GC content across all chromosomes varied from 38% (chromosome 5) to 48%(chromosome 19), with an average of 41% (Supplementary Table S5).
Compared to the other reportedMacacagenomes, the Tibetan macaque genome was the most contiguous, with the least number of contigs (893) and scaffolds (624) and the shortest lengths of gaps and unplaced scaffolds(Supplementary Table S4). Genome alignment of long and short reads showed high mapping (long reads: 92.45%; short reads: 99.97%) and coverage rates (long reads: 99.97%; short reads: 99.96%), indicating high consistency between the reads and genome assembly (Supplementary Table S6). Of the 13 780 primate orthologs, 13 172 (95.6%) were identified as complete BUSCO genes, 1.2% as partial BUSCO genes,and 3.2% as missing BUSCO genes. BUSCO completeness of the Tibetan macaque genome was the highest among all sequenced macaque genomes (Figure 1D). Thus, the quality of the assembly was highest with respect to continuity and completeness among all available macaque genome assemblies.
Repetitive sequences were annotated using RepeatMasker(v4.1.2) and RepeatModeler (v2.0.1), with repetitive elements found to constitute 47.14% of the Tibetan macaque genome(Supplementary Table S7). Among transposable elements(TEs), retroelements, consisting of long interspersed elements(LINEs, 21.74%), short interspersed elements (SINEs,11.10%), and long terminal repeats (LTRs, 9.07%), occupied 41.92% of the genome. There were 13 102 repetitive elements located in unplaced contigs, totaling 8 297 399 bp and accounting for 69.82% of all unplaced contigs. The high repeat content of these contigs may help explain the difficulty of placing them on chromosomes. In addition, the content and composition of the repetitive elements were similar among the six macaque genomes and no species-specific TE family was found (Supplementary Table S8 and Figure S1).
Homology annotation, transcriptome annotation, andab initioprediction were used in combination for gene prediction of the Tibetan macaque genome. A final set of 22 485 genes were annotated in the genome (Supplementary Table S9).Gene length ranged from 150 bp to 835 594 bp, with an average of 8.53 exons per gene and average coding sequence (CDS) length of 176.48 bp per exon. Average gene and CDS lengths were 39 883.31 and 1 505.48 bp,respectively. There were 4 662 single-exon genes, with exon length ranging from 150 to 6 102 bp. A total of 22 128 genes(98.41%) were located on chromosomes and only 357 genes(1.59%) were located on unplaced scaffolds. Chromosome 1 contained the most genes (2 065), while chromosome Y contained the least (121). Chromosomal gene density ranged from 4.61 genes/Mb (chromosome 17) to 20.80 genes/Mb(chromosome 19) (average genome-wide density of 7.97 genes/Mb) and gene density was positively associated with GC content (correlation value: 0.73;P-value: 0.0)(Supplementary Table S5; Figure 1C).
Protein-coding genes were aligned against several known public databases, including Swiss-Prot, TRanslation of EMBL(TrEMBL), NCBI nonredundant protein sequences (NR),InterProScan, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG). Finally, 21 870 (97.26%)protein-coding genes were annotated based on at least one of the databases. Most genes (17 716, 78.79%) were annotated in all six databases, indicating a highly credible gene set(Supplementary Table S10 and Figure S2).
Non-coding RNAs (ncRNAs) were annotated using different methods. For transfer RNAs (tRNAs) and ribosomal RNAs(rRNAs), we used tRNAscan-SE (v2.0.9) and RNAmmer(v1.2) with default parameters, respectively. For other ncRNAs, such as long ncRNAs (lncRNAs), microRNAs(miRNAs), and small nuclear RNAs (snRNAs), annotations were performed by searching the Rfam database (v14.4)using INFERNAL (v1.1.2). We predicted 450 tRNA genes,2 488 miRNA genes, 3 214 snRNA genes, 135 rRNA genes,and 211 lncRNA genes, covering 0.24% of the genome(Supplementary Table S11).
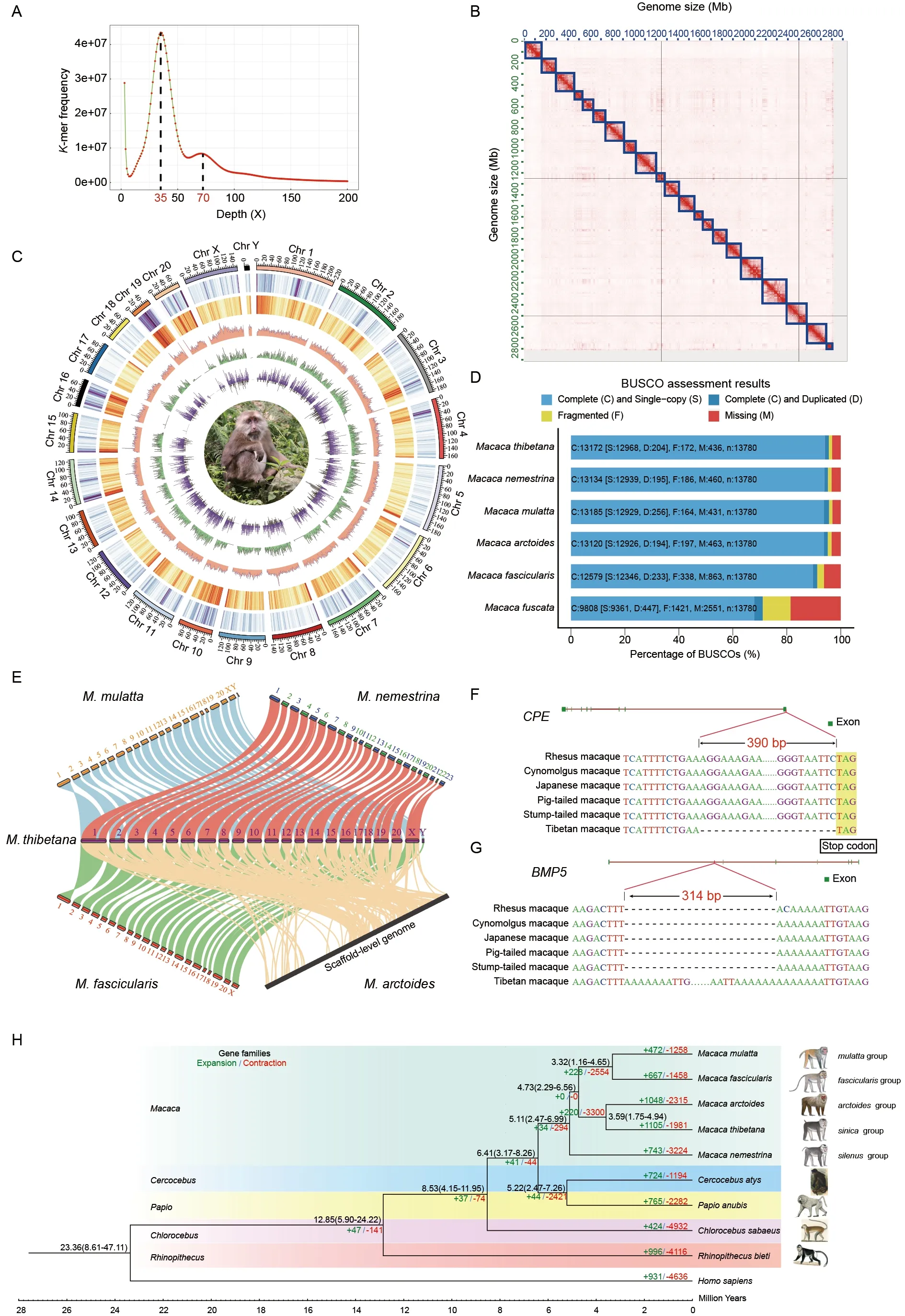
Figure 1 Genome analysis of Tibetan macaque
Syntenic relationships among the sequenced macaque genomes were analyzed and plotted using MCScan (Python).Substantial genome-wide collinearity was revealed between the Tibetan macaque and other four macaque species(Figure 1E). The Tibetan macaque chromosomes displayed a one-to-one corresponding relationship with the rhesus and cynomolgus macaque chromosomes, with reverse complementary sequence relationships also clearly observed in several chromosomes, e.g., chromosome 1 (Supplementary Figure S3A). Some errors caused by assembly using Hi-C data were observed (Supplementary Figure S3B), but no chromosome rearrangement was found. Although a good syntenic relationship was observed between the Tibetan and pig-tailed macaque assemblies, there were several errors,e.g., some contigs on chromosomes 1, 2, and 19 were not anchored on the corresponding superscaffolds between the two assemblies. Furthermore, superscaffolds 10 and 11 in the pig-tailed macaque assembly should correspond to chromosome 10, and superscaffolds 22 and 23 should correspond to chromosome X. In addition, although the stumptailed macaque genome was at the scaffold level, it appeared to be nearly complete without an obvious lack of fragments.Thus, genomic syntenic analysis indicated that the Tibetan macaque genome assembly was of high quality and gene arrangements were highly conserved among macaque species.
Gene families in the Tibetan macaque and other primates(i.e., rhesus macaques, cynomolgus macaques, pig-tailed macaques, stump-tailed macaques, olive baboons, black snub-nosed monkeys, African green monkeys, sooty mangabeys, and humans) were identified using OrthoFinder(v2.3.11) with default settings. A total of 29 495 orthogroups were detected, of which 8 178 contained one-to-one orthologous genes. These genes were aligned using MAFFT(v7.475) with default parameters, and a phylogenetic tree was constructed using maximum-likelihood in RAxML (v8.2.4) with the GTRGAMMA model. Divergence time between species was estimated using MCMCTREE in the PAML (v4.8)package. We used two calibrations for analysis: (1)Divergence time betweenM. mulattaandM. fasciculariswas set to 2.56-5.11 Ma (http://www.timetree.org/); and (2)Divergence time between thePapioandMacacagenera was set to 5-8 Ma (Roos et al., 2019). The phylogenetic tree indicated that Tibetan macaques were most closely related to the stump-tailed macaques, rhesus macaques were grouped with cynomolgus macaques, and pig-tailed macaques diverged first from the other macaque species (Figure 1H).These phylogenetic relationships are consistent with previous studies based on nuclear genes and resequencing data (Fan et al., 2018; Song et al., 2021), with 100% bootstrap support for every branch. Divergence time estimation suggested that theMacacagenus diverged from thePapiogenus approximately 6.41 Ma, and pig-tailed macaques and other macaques diverged about 5.11 Ma. The Tibetan and stumptailed macaques diverged about 3.59 Ma (Figure 1H). These divergence times are consistent with previous study based on mitochondrial genomes (Roos et al., 2019). However, our estimation was slightly older compared to other studies based on single nucleotide variations among macaque genomes(Fan et al., 2018; Song et al., 2021), which could be attributed to different molecular genetic markers.
Based on the identified gene families and phylogenetic tree with predicted species divergence times, CAFÉ (v4.2) was used to predict gene family expansions and contractions.Results revealed 1 105 expanded and 1 981 contracted gene families in the Tibetan macaque genome (Figure 1H). Both GO and KEGG functional enrichment analysis of the expanded gene families was performed using the R package clusterProfiler and KOBAS v3.0 with an adjustedP-value of 0.05. The expanded gene families were mainly related to biosynthesis and metabolism (Supplementary Tables S12,S13; Supplementary Figure S4). For example, we found an increase in the copy number of theGAPDH(glyceraldehyde-3-phosphate dehydrogenase) gene involved in the glycolytic process (GO: 0006096) and glucose metabolic process (GO:0006006) (Supplementary Table S14), which may be related to the comparatively large body size of Tibetan macaques.
The alignments of one-to-one orthologous genes and phylogenetic tree were used to estimate the ratio of nonsynonymous to synonymous substitutions per gene using maximum-likelihood with the codeml program in PAML (v4.8)under the branch-site model. Potential false-positive PSGs were filtered and removed based on the following criteria:PSGs with continuous positively selected sites (PSSs), PSGs with ≥ 3 PSSs within 10 bp, and PSGs with PSSs at the first or last position of amino acid sequences. We found that 454 of the 8 178 one-to-one orthologous genes were under positive selection in the Tibetan macaque. Based on functional enrichment analysis, genes involved in protein binding (GO:0005515), anatomical structure development (GO: 0048856),and developmental process (GO: 0032502) were significantly enriched in the PSGs (Supplementary Table S15;Supplementary Figure S5), with several of these PSGs likely related to large body size.
Tibetan macaques have a shorter tail and larger body size compared to other macaque species. Notably, four genes associated with tail development and eight genes associated with body size were under positive selection (Supplementary Table S16). For example, a Tibetan macaque-specific mutation (Ala100Thr; PROVEAN score: 0.064; Neutral) and a macaque-specific mutation (Asp279Glu) were found in theBMPER(bone morphogenetic protein-binding endothelial cell precursor-derived regulator) gene (Supplementary Figure S6A).BMPERis involved in organogenesis and morphogenesis through the bone morphogenetic protein(BMP) signaling pathway (Ikeya et al., 2006). Furthermore,BMPERknockout mice present with vertebral and cartilage development abnormalities, eye malformations, renal hypoplasia, and alveolar lung defects (Ikeya et al., 2006). We also identified a specific insertion in theKATNB1gene of Tibetan and stump-tailed macaques (Supplementary Figure S6B), indicating that this mutation may have occurred before divergence of the two species. ThePTTNgene contained three Tibetan macaque-specific mutations, all predicted to be neutral by PROVEAN, as well as a Tibetan and stump-tailed macaque-specific mutation and five macaque-specific mutations (Supplementary Figure S6C). Positive selection analysis suggested that the PSGs may play important roles in the Tibetan macaque phenotype and adaptations.
The whole-genome sequences of five macaque species(rhesus, cynomolgus, pig-tailed, stump-tailed, and Japanese macaques) were aligned with the Tibetan macaque genome sequences and TMSSVs were identified using MUM&Co.TMSSV regions were defined as intersecting based on the following criteria: (1) SVs were identified in all five Tibetan macaque-other macaque pairwise comparisons; (2)Overlapping region length of five SVs was longer than half of the shortest SV; and (3) SVs located in unplaced scaffolds were excluded. TMSSVs located in genes were searched using bedtools (v2.30.0), and genes related to tail development and body size were searched in the published literature and MGI databases (http://www.informatics.jax.org/)based on their known functions.
Based on sequence alignments of the Tibetan macaque and other five macaque species, deletions and insertions accounted for a large proportion of SVs detected(Supplementary Table S17). After filtering, we identified 6 788 candidate TMSSVs, including 5 862 insertions, 920 deletions,one tandem contraction, and five tandem duplications(Supplementary Figure S7A). In total, TMSSVs covered 7 489 362 bp, accounting for 0.27% of the Tibetan macaque genome. Insertion lengths ranged from 65 bp to 105 586 bp,with a total of 5.58 Mb. Deletion lengths ranged from 88 bp to 58 272 bp, with a total of 1.91 Mb. Median lengths for insertions and deletions were 317 bp and 1 034 bp,respectively (Supplementary Figure S7B). The TMSSVs were not equally distributed across the genome, with lower densities on chromosomes 16 and 19 (Figure 1C).Furthermore, 2 050 (31.2%; 1 802 insertions, 244 deletions,and four duplications) TMSSVs partially or entirely overlapped protein-coding genes, and the remaining 4 738 (69.8%; 4 060 insertions, 676 deletions, one contraction, and one duplication) were located in the intergenic regions.
Among the 2 050 TMSSVs overlapping protein-coding genes, only 37 overlapped exons, including 29 insertions and eight deletions, resulting in mutations in 41 genes(Supplementary Table S18). A 390 bp deletion specific to the Tibetan macaque genome was found near the stop codon of the last exon ofCPE(Figure 1F; Supplementary Table S19).CPEis an obesity susceptibility gene, and mice harboring homozygous alleles of a naturally occurring loss-of-function mutation ofCPEare obese, infertile, and diabetic (Naggert et al., 1995). We also identified two deletions and four insertions located in introns of five genes (BMP5,ARHGAP18,ARID1B,B2W2, andCOL9A2) related to tail development or body size(Supplementary Table S19). A 314 bp insertion inBMP5was specific in the Tibetan macaque genome (Figure 1G).BMP5induces bone and cartilage development, differentiates osteoprogenitor mesenchymal cells, and up-regulates osteoblastic features to influence cytokines and growth factors(Sykaras & Opperman, 2003). Mutations inBMP5are associated with a wide range of skeletal defects, such as reduction in long bone width and vertebral process size, as well as a reduction in body mass (Kingsley et al., 1992).However, the exact effects of these TMSSVs and their molecular mechanisms require further functional validation to confirm the association between TMSSVs and Tibetan macaque phenotypes.
In this study, we assembled the first chromosome-level genome of the Tibetan macaque and performed associated annotations. This assembly will expand macaque genome resources and provide a molecular basis for studying the adaptation and evolution of Tibetan macaques.
DATA AVAILABILITY
The raw read data, Hi-C data, and final genome assembly were deposited in the CNGB Sequence Archive (CNSA) of the China National GeneBank Database (CNGBdb)(https://db.cngb.org/) under project ID CNP0002784 and the National Center for Biotechnology Information (NCBI)database under BioProjectID PRJNA821381. The genome assembly (fasta) was also deposited in the Science Data Bank(https://www.scidb.cn/s/iIvEfq) under DOI:10.57760/sciencedb.j00139.00023 and NGDC(https://ngdc.cncb.ac.cn/) under project PRJCA010663.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
J.L. led the project. R.Z., Z.F., and J.L. designed the study.R.Z., C.Z., X.J., and K.L. performed bioinformatics analysis.R.Z. analyzed the results and wrote the manuscript with input from the other authors. J.L., C.Z., and J.X. revised the article.All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are very grateful to Megan Price for English correction and proofreading. We sincerely thank Sichuan Hengshu Biotechnology Co., Ltd. (Yibin, China) for providing macaque samples. We thank Dr. Jianming Zeng (University of Macau)and all members of his bioinformatics team at Biotrainee for generously sharing their experience and codes. We thank Shanghai Hengsheng Biotechnology Co., Ltd. for providing high-performance computing cluster (https://biorstudio.cloud).
Ru-Song Zhang1,#, Chuang Zhou1,2,#, Xian-Lin Jin1, Kang-
Hua Liu1, Zhen-Xin Fan1,2, Jin-Chuan Xing3, Jing Li1,2,*1Key Laboratory of Bio-Resources and Eco-Environment (Ministry
of Education),College of Life Sciences,Sichuan University,Chengdu,Sichuan610065,China2Sichuan Key Laboratory of Conservation Biology on Endangered
Wildlife,College of Life Sciences,Sichuan University,Chengdu,Sichuan610065,China3Department of Genetics and Human Genetics Institute of New
Jersey,Rutgers,State University of New Jersey,Piscataway
08854,USA
#Authors contributed equally to this work*Corresponding author, E-mail: ljtjf@126.com

- Zoological Research的其它文章
- Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
- The Hippo pathway and its correlation with acute kidney injury
- Genomics and morphometrics reveal the adaptive evolution of pikas