
Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
2022-10-17 03:27DalangYu,JunweiZhu,JianingYang等
Zoological Research 2022年5期
DEAR EDITOR,
The COVID-19 pandemic caused by SARS-CoV-2 continues to pose a tremendous threat to human society. SARS-CoV-2 is airborne and transmits primarily through social contact;however, whether cold chain-related transmission has occurred remains highly debated (Han & Liu, 2022; Lewis,2021; Ma et al., 2021; Mallapaty et al., 2021; Pang et al.,2020; Wu et al., 2021). Here, we present a novel method and identify two transmission routes based on lineage-specific reductions in the SARS-CoV-2 evolutionary rate. After analyzing 4 039 521 SARS-CoV-2 genomic sequences, we identified two outbreaks in Xinfadi-Beijing and Auckland that may be cold-chain related, caused by two mutation-dormant variants, respectively. A mutation-dormant variant represents a variant with a (nearly) identical genomic sequence repeatedly isolated over a long period of time. Dalian outbreak in July 2020 and Yingkou outbreak ten months later were epidemiologically connected and derived from a mutationdormant variant. Furthermore, the earliest SARS-CoV-2 variant (i.e., the most recent common ancestor of SARS-CoV-2) was also found to be a mutation-dormant variant. Its spillover events were repeatedly observed during the last two years, indicating that the COVID-19 outbreak in Wuhan may be associated with spillover events from coldchains. In all observed cases, the virus re-started mutating after spillover events from cold-chains. Systematic identification of spillover events revealed that the frequency of cold-chain related transmission is in the order of 0.1%-10%.Our results indicate that that cold-chain related transmission is rare but may have occurred globally.
Cold-chain related SARS-CoV-2 transmission may be possible because the virus can remain infectious but inactive under low temperatures and suitable humidity. For example,the virus shows little reduction in infectivity when maintained at 4 °C for 14 days (Chin et al., 2020). However, many key factors must be satisfied to allow cold-chain related SARSCoV-2 transmission, such as viral load on the contaminated surfaces, contaminated areas, environmental conditions during transportation, and amount of virus to which a person is exposed. These factors are highly variable, and it is not ethically possible to test these factors using a population cohort. Therefore, a novel approach is needed to analyze realworld examples and examine whether cold-chain related SARS-CoV-2 transmission has occurred. A large number of viral genomic sequences and their epidemiologically associated metadata are available, which may contain essential information about viral transmission.
In this study, we used the Coronavirus GenBrowser (CGB)platform (Yu et al., 2022), which provides a panoramic view of the transmission and evolution of SARS-CoV-2, to examine available molecular epidemiological evidence. All studied SARS-CoV-2 strains were isolated from humans, with each sequence representing a strain isolated from a patient, and those sequences representing viral transmission outcome.The genome-wide evolutionary rate was estimated to be 9.69×10-4per site per year (95% confidence interval:7.27×10-4-1.23×10-3) or 7.88×10-2per genome per day,similar to that reported in other studies (Nie et al., 2020; Yu et al., 2020) but obtained with a much larger sample size.Different evolutionary rates were obtained for different genes(Supplementary Table S1). Evolutionary rate heterogeneity among different viral genes was expected, as observed in other organisms.
When SARS-CoV-2 is transmitted through social contact,the virus duplicates in human hosts with a certain error rate(i.e., mutation rate). In this process, mutations are inevitable if the considered duration is long (Figure 1A). In contrast, the virus does not replicate and mutate under cold-chain conditions. The mutation-dormant virus can be detected in the CGB dataset after spillover events from the cold-chain. If the considered duration is long, two routes of transmission can be distinguished by detecting whether SARS-CoV-2 evolves significantly slowly in the lineage. As expected, the virus restarts mutating after spillover events from cold-chains.Moreover, repeated spillover events may affect the distribution of the evolutionary rate of the sample (Figure 1B). When there is no cold-chain related transmission, the simulated distribution of evolutionary rate is unimodal. However, the observed distribution of evolutionary rate appears to be bimodal. Therefore, the cold-chain related transmission has a visible effect on the evolutionary rate of the sample.
The new method was validated by analyzing two outbreaks for which epidemiological information is available, i.e.,outbreak in Xinfadi (Beijing, China) in June 2020(Supplementary Figure S1) and outbreak in Auckland (New Zealand) after 102 days of no new locally transmitted COVID-19 cases (Supplementary Figure S2). While there are no official reports on whether Auckland outbreak was related to cold-chain, the first person who reported illness onset was an employee at a cool storage facility. In both cases, mutationdormant variants were identified, suggesting that both outbreaks may have resulted from spillover events from coldchains (see Supplementary Materials). Therefore, pandemicscale phylogenomics can be used to reliably detect global cold-chain related SARS-CoV-2 transmission.
There was a COVID-19 outbreak in Dalian in July 2020 and a multi-city outbreak originating in Yingkou in May 2021(Supplementary News 1, 2). Dalian outbreak began with workers handling contaminated cold-chain pollock packages from an overseas cargo ship (Ma et al., 2021). In Yingkourelated outbreak, the first case was found in Lu’an, with multiple cases emerging in multiple cities thereafter, all of which were social contacts of confirmed cases in Yingkou(Figure 1C). For this outbreak, all genomic sequences in public databases were collected from Lu’an. Thus, Yingkourelated outbreak was represented by the Lu’an samples in the evolutionary tree (Figure 1D). The two outbreaks were considered independent as no connected cases were identified over 300 days. CGB lineage tracing showed that the sequences of three environmental samples collected from pollock packages in Dalian in May and June 2020 were identical to that of the most recent common ancestor (MRCA)of cases in the two outbreaks. The Lu’an samples derived only three mutations over 294 days, with a significantly reduced evolutionary rate (P=2.03×10-7) after Bonferroni correction(Dunn, 1961) of multiple tests (n=7). TheP-value was 4.44×10-9when evolutionary rate heterogeneity was considered. The conclusion remained the same even when using the lower bound of the confidence interval of the estimated evolutionary rate (P=2.93×10-5). These results suggested that the two outbreaks may be epidemiologically connected via a mutation-dormant variant, and Yingkourelated outbreak is most likely cold-chain related.
We used the latest CGB global dataset (data.2022-06-03,n=4 039 521 genomic sequences) to examine whether other spillover events from these contaminated cold-chain products occurred outside of China. However, we did not find any new spillover events.
As pandemic-scale phylogenomics analysis can reliably detect cold-chain related transmission, this method can be used to reveal previously unknown cold-chain related transmission. The reference genomic sequence Wuhan-Hu-1(GenBank accession number: NC_045512) (Wu et al., 2020)represents the MRCA sequence of the earliest SARS-CoV-2 variant (Yu et al., 2022). Therefore, we examined whether Wuhan-Hu-1 is a mutation-dormant variant, which is essential for studying the origin of SARS-CoV-2.
Among the 1 610 125 high-quality genomic sequences, we identified 250 strains with genomic sequences identical to Wuhan-Hu-1 (Figure 1E; Supplementary Table S2). These strains were collected from 32 countries/regions between December 2019 and August 2021. Most of these non-mutated strains (177/250=70.8%) were collected in Asia and North America, but the highest frequency was observed in Africa(Supplementary Figure S3). In this dataset, the two most recent strains were collected from New York in June 2021 and Denmark in August 2021, indicating that the variant did not mutate in the lineage within 20 months. Thus, the variant showed a significantly reduced evolutionary rate in the lineage(P=7.44×10-22) after Bonferroni correction (Dunn, 1961) for multiple tests (n=1 610 125). Even with the lower bound confidence interval (P=1.41×10-16), the conclusions remained the same, indicating that our finding is robust with the estimated evolutionary rate of SARS-CoV-2.
The reduced evolutionary rate was a lineage-specific effect.The mutation-dormant variant re-started mutating once the virus re-infected a human host (i.e., a spillover event from a cold-chain) and was transmitted through social contact. Three spillover clades from cold-chains were presented, i.e., humanto-human transmission lineages identified through genome sampling in Germany (sub-panel of Figure 1E), the UK(Supplementary Figure S4A) and the USA (Supplementary Figure S4B). Sequences identical to Wuhan-Hu-1 were found in 32 countries/regions and sequenced by different institutes,so contamination caused by Wuhan-Hu-1 strain samples is an unlikely explanation. Moreover, contamination cannot explain the observed mutations in the genomic sequences after spillover events from cold-chains (Figure 1E, Supplementary Figure S4).
We further examined the latest CGB global dataset(data.2022-06-03,n=4 039 521 genomic sequences) and identified more recent spillover events. While the quality of early viral sequences was suboptimal, this did not affect our findings as the test depends on the time difference between the collection date of the reference genome and that of the recent identical genomes. Therefore, the earliest SARS-CoV-2 variant is mutation-dormant, suggesting that its origin and spread may be cold-chain related.
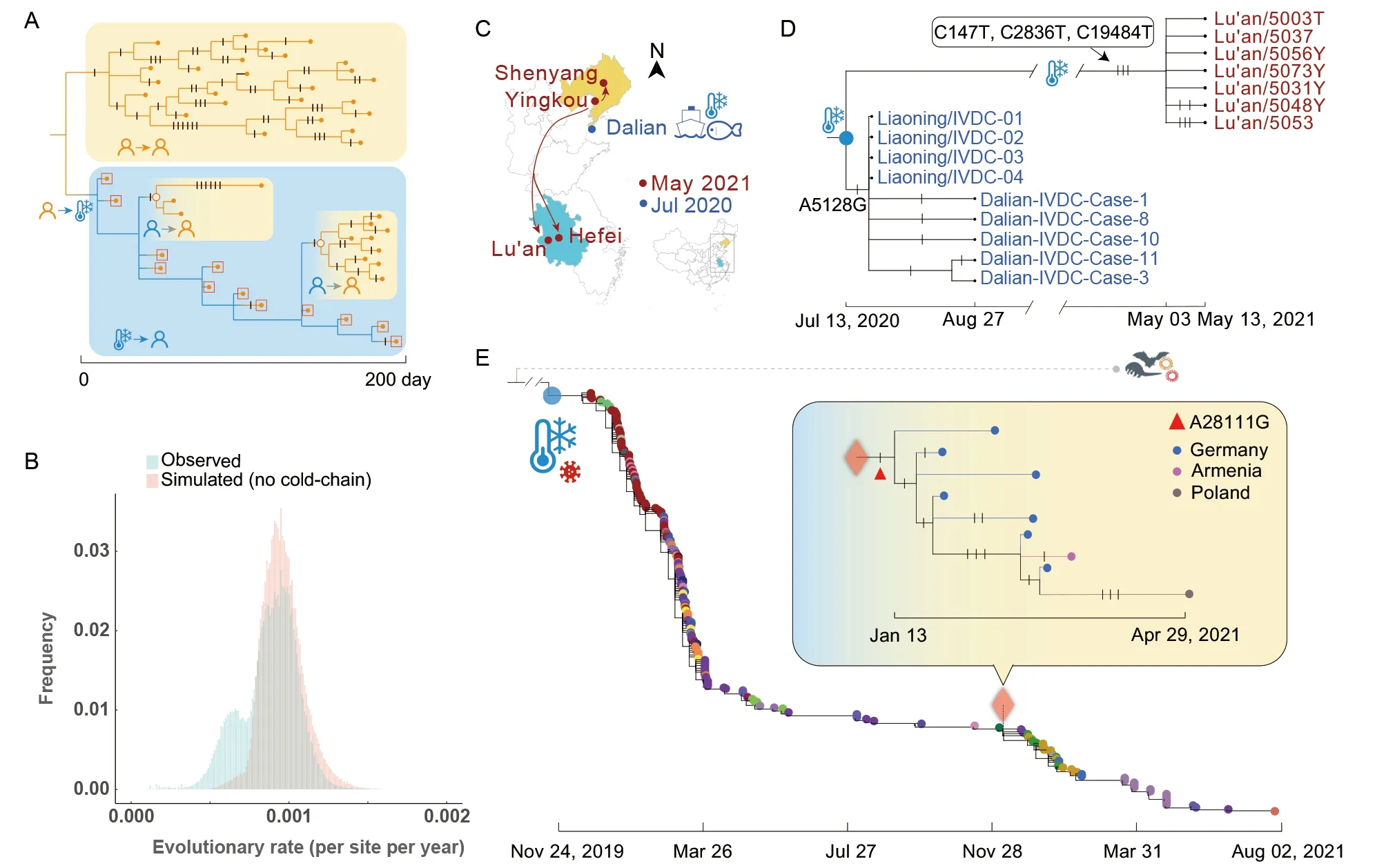
Figure 1 Identification of cold-chain related SARS-CoV-2 transmission using pandemic-scale phylogenomics
Additional mutation-dormant variants and putative spillover events can be identified by pandemic-scale phylogenomics,such as the putative cold-chain related spread of D614G variant and Delta variant of concern (VOC) (Supplementary Figures S5, S6). Thus, we estimated the number of mutationdormant variants by scanning the massive SARS-CoV-2 tree with 1 610 125 high-quality genomic sequences. We identified 362 mutation-dormant variants that did not mutate within more than 100 days (Supplementary Table S3). This small number was expected because the occurrence and identification of a mutation-dormant variant requires contaminated cold-chain products, repeated spillovers, and identification of spillovers at different time points.
By counting the identical descendants of those variants, we found 60 796 strains in total. Among these putatively coldchain related strains, 317 were collected from South America,6 053 from Asia, 42 935 from Europe, 244 from Africa, 11 080 from North America, and 167 from Oceania, indicating that cold-chain related transmission may have occurred in all inhabited continents. The percentage of cold-chain related strains was not high among total samples (60 796/1 610 125=3.78%) and was similar in different continents (i.e., 2.42%,4.94%, 4.08%, 3.71%, 2.73%, and 1.62%, respectively).Therefore, these results indicate that the frequency of coldchain related transmission is rare.
In conclusion, we developed a pandemic-scale phylogenomics method to detect cold-chain related SARSCoV-2 transmission. The method has been implemented in the CGB (Yu et al., 2022) and can identify newly emerged cold-chain related transmissions. Cold-chain related transmission may have happened globally but appears to be rare, thus making human society relatively easier to control the transmission. Moreover, proper protection and training of workers in cold-chain facilities is important, not only to protect workers from cold-chain related infection, but also to prevent potential spread from infected workers. Educating consumers on how to properly handle cold-chain products produced in epidemic areas would also be helpful. Well-established coldchains are essential for the economy and global distribution of food and drink. Therefore, the prevention of cold-chain related transmission is necessary to reduce the impact of the pandemic on global cold-chains and avoid cold-chain associated adaptation of SARS-CoV-2 in the future.
DATA AND SOFTWARE AVAILABILITY
Data were downloaded from https://ngdc.cncb.ac.cn/ncov/apis/ and the CGB, as a free plug-in of the eGPS Desktop (Yu et al., 2019), is available from http://www.egps-software.net/egpscloud/eGPS_Desktop.html.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Conceptualization: D.Y., J.Z., J.Y., Y.H.P., G.P.Z., Y.P.Z,W.Z., G.Z., and H.L.; Coding and software development: D.Y.,J.Y., H.M., Z.Q.H., and L.D.; Data integration: D.Y., J.Z., J.Y.,R.C., B.T., and G.D.; Data analysis: D.Y., J.Z., J.Y., and Y.H.P.; Writing: D.Y., J.Y., Y.H.P., G.P.Z., Y.P.Z, W.Z., G.Z.,and H.L.; Supervision & funding acquisition: Y.H.P., G.P.Z.,Y.P.Z, W.Z., G.Z., and H.L. All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge the researchers who generated and deposited SARS-CoV-2 sequence data in GISAID,GenBank, CNGBdb, GWH, and NMDC.
Dalang Yu1,8,#, Junwei Zhu2,#, Jianing Yang1,8,#,Yi-Hsuan Pan3,#, Hailong Mu1, Ruifang Cao1, Bixia Tang2,Guangya Duan2,8, Zi-Qian Hao1,8, Long Dai1,7,Guo-Ping Zhao1,5,6, Ya-Ping Zhang4, Wenming Zhao2,8,*,Guoqing Zhang1,8,*, Haipeng Li1,8,*1National Genomics Data Center&Bio-Med Big Data Center,CAS Key Laboratory of Computational Biology,Shanghai Institute of Nutrition and Health,Chinese Academy of Sciences,Shanghai
200031,China
2National Genomics Data Center,Beijing Institute of Genomics,Chinese Academy of Sciences and China National Center for Bioinformation,Beijing100101,China
3Key Laboratory of Brain Functional Genomics of Ministry of Education,School of Life Science,East China Normal University,Shanghai200062,China
4State Key Laboratory of Genetic Resources and Evolution,Yunnan Laboratory of Molecular Biology of Domestic Animals,Kunming Institute of Zoology,Chinese Academy of Sciences,Kunming,Yunnan650223,China5Key Laboratory of Synthetic Biology,CAS Center for Excellence
in Molecular Plant Sciences,Institute of Plant Physiology and Ecology,Chinese Academy of Sciences,Shanghai200032,China
6School of Life and Health Sciences,Hangzhou Institute for Advanced Study,University of Chinese Academy of Sciences,Hangzhou,Zhejiang310024,China7Shanghai Southgene Technology Co. Ltd,Shanghai201203,China8University of Chinese Academy of Sciences,Beijing100049,China
#Authors contributed equally to this work*Corresponding authors, E-mail: zhaowm@big.ac.cn;gqzhang@picb.ac.cn; lihaipeng@picb.ac.cn

- Zoological Research的其它文章
- Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- The Hippo pathway and its correlation with acute kidney injury
- Genomics and morphometrics reveal the adaptive evolution of pikas
- Molecular phylogeny and taxonomy of four Remanella species (Protozoa, Ciliophora): A flagship genus of karyorelictean ciliates, with descriptions of two new species