
The Hippo pathway and its correlation with acute kidney injury
2022-10-17 03:27ChiZhangChuanLeiLiKeXinXuZhiHuangZhengGuoZheChengHuiJuanWuJunLiu
Zoological Research 2022年5期
Chi Zhang, Chuan-Lei Li, Ke-Xin Xu, Zhi-Huang Zheng, Guo-Zhe Cheng, Hui-Juan Wu, Jun Liu,*
1 Department of Nephrology, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 201600, China
2 Department of Pathology, School of Basic Medical Science, Fudan University, Shanghai 200030, China
ABSTRACT Acute kidney injury (AKI) is a significant clinical complication with a substantial impact on morbidity and mortality, for which therapeutic options remain limited. The Hippo signaling pathway is an evolutionarily conserved pathway implicated in cell proliferation, dedifferentiation, and apoptosis via phosphorylation and inactivation of its downstream effectors Yes-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif(TAZ). Recent studies have revealed that the Hippo pathway plays a pivotal role in the pathogenesis and repair of AKI. The Hippo pathway can mediate renal dysfunction through modulation of mitochondrial apoptosis under AKI conditions. Transient activation of YAP/TAZ in the acute phase of AKI may benefit renal recovery and regeneration, whereas persistent activation of YAP/TAZ in severe AKI may lead to maladaptive repair and transition to chronic kidney disease. This review aims to summarize recent findings on the associations between the Hippo pathway and AKI and to identify new therapeutic targets and strategies for AKI.
Keywords: Hippo pathway; Acute kidney injury(AKI); Maladaptive repair; Fibrosis; Chronic kidney disease (CKD)
lNTRODUCTlON
Acute kidney injury (AKI) remains a major medical challenge with high morbidity, mortality, and healthcare costs (Zuk &Bonventre, 2019). Apart from supportive treatment, few preventive and therapeutic options currently exist (Lameire et al., 2013). Among survivors, long-term outcomes of AKI include development of chronic kidney disease (CKD), endstage renal disease (ESRD), and death (Lameire et al., 2013;Venkatachalam et al., 2015; Zuk & Bonventre, 2019).Incomplete recovery from AKI, also called maladaptive repair,can lead to long-term functional deficits and CKD. The progression of adaptive to maladaptive kidney repair in AKI leading to CKD can be attributed to G2/M cell cycle arrest,profibrotic cytokine secretion, myofibroblast generation,extracellular matrix (ECM) production, and inflammation(Ferenbach & Bonventre, 2015; Yu & Bonventre, 2020).
The mammalian Hippo pathway is a serine/threonine kinase cascade composed of mammalian sterile 20-like 1/2(MST1/2), large tumor suppressor homolog 1/2 (LATS1/2),and Yes-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif (TAZ) (Zhao et al., 2011b). It regulates cell growth and fate determination, organ size, and regeneration through the phosphorylation and inactivation of its downstream effectors YAP/TAZ (Ma et al., 2019). Recent studies have implicated the Hippo pathway in the pathogenesis and repair of AKI and transition to CKD. In this review, we summarize current findings on the correlations between the Hippo pathway and kidney injury and shed some light on future treatment for acute or chronic kidney diseases.
HlPPO PATHWAY lN MAMMALS: A KlNASE CASCADE
The Hippo pathway is highly evolutionarily conserved in mammals and consists of a three-step kinase cascade of MST1/2 (also called STK4/3), LATS1/2, and YAP/TAZ (also called WWTR1). Upon Hippo pathway activation, Salvador homolog 1 (Sav1) interacts with and is phosphorylated by MST1/2, thereby acting as a partner of MST1/2 to promote phosphorylation of LATS1/2 (Callus et al., 2006; Tapon et al.,2002). LATS1/2 are activated by MST1/2 through multiple mechanisms: MST1/2 can directly phosphorylate LATS1/2 at the C-terminal hydrophobic motif (Thr1079 for LATS1 and Thr1041 for LATS2), promoting LATS1/2 autophosphorylation at its activation loop. Furthermore, MST1/2-phosphorylated Mps one binder 1 (MOB1) binds to the autoinhibitory domain of LATS1/2, leading to full activation of LATS1/2 (Callus et al.,2006; Chan et al., 2005; Praskova et al., 2004; Wu et al.,2003). Neurofibromin 2 (NF2) can also directly interact with and recruit LATS1/2 to the plasma membrane for phosphorylation by MST1/2 (Yin et al., 2013; Zhang et al.,2010). Activated LATS1/2 then phosphorylate serine on the YAP/TAZ HXRXXS motifs (Hao et al., 2008; Lei et al., 2008;Liu et al., 2010; Zhao et al., 2007, 2010). Phosphorylation of YAP Ser127 and TAZ Ser89 generates 14-3-3 binding sites,and binding to 14-3-3 results in cytoplasmic sequestration(Dong et al., 2007; Zhao et al., 2007). In addition,phosphorylation of YAP Ser381 and TAZ Ser311 leads to subsequent phosphorylation by casein kinase 1 (CK1) and activation of phosphodegron, resulting in the recruitment of SCFbeta-TRCP E3 ubiquitination ligase and proteasomal degradation of both YAP and TAZ (Liu et al., 2010; Zhao et al., 2010).
When upstream kinases of the Hippo pathway are inactivated, YAP/TAZ are dephosphorylated and activated. As transcriptional co-activators, YAP/TAZ enter the nucleus and bind with TEAD family transcription factors (TEAD 1-4) to induce the expression of target genes (Zhao et al., 2008),such asCTGF,Cyr61,ANKRD1,DIAPH3,amphiregulin, andsurvivin, thereby improving cell proliferation and inhibiting cell death (Bai et al., 2012; Rinschen et al., 2017). Vestigial-like family member 4 (VGLL4) competes with YAP/TAZ for TEAD binding in the nucleus and represses target gene expression(Figure 1) (Jiao et al., 2014; Zhang et al., 2014). The association between the Hippo pathway and AKI has attracted increasing attention, and the effects of Hippo pathway component activation on AKI are summarized in Table 1.
HlPPO PATHWAY REGULATlON

Figure 1 Hippo pathway in mammalian cells
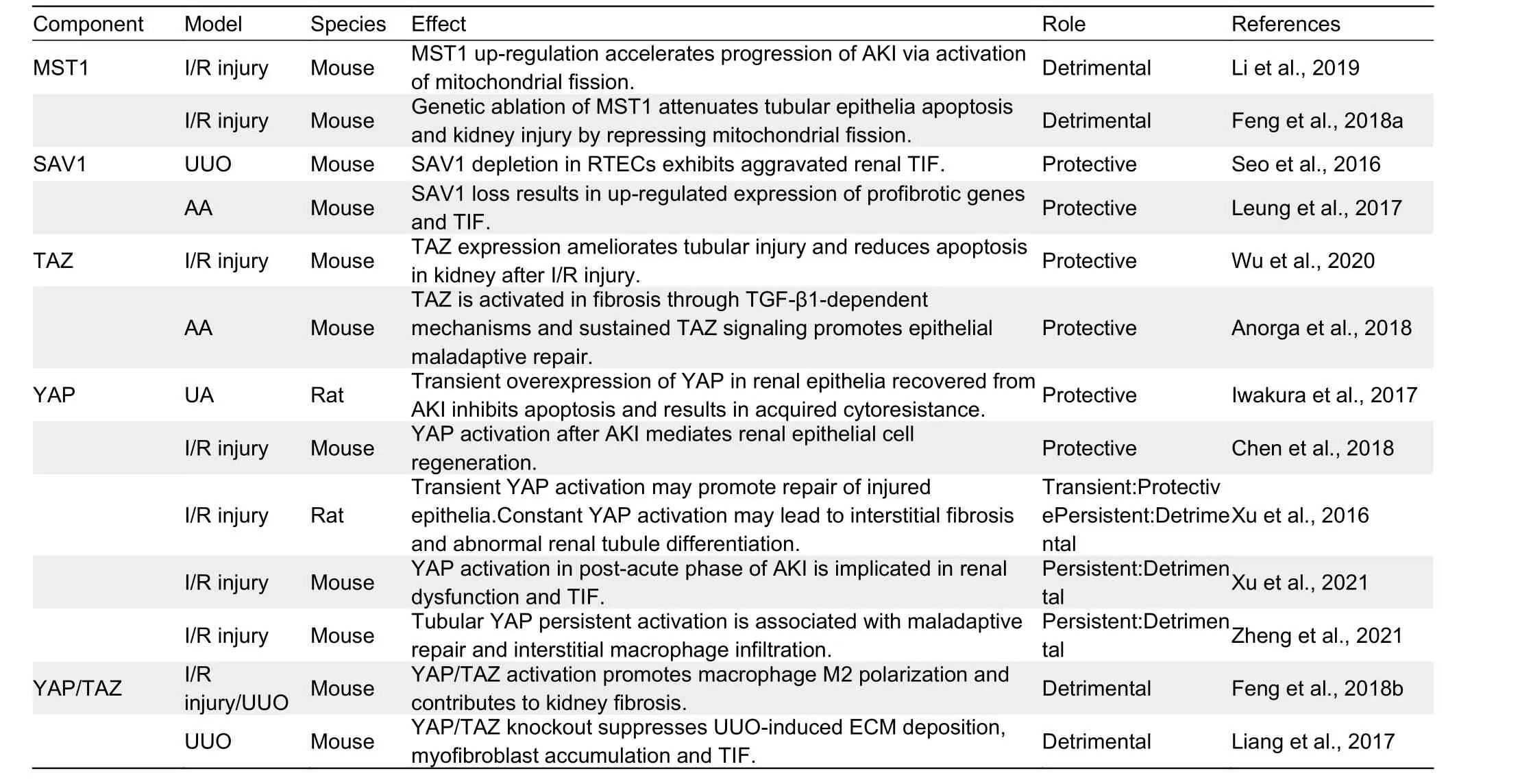
Table 1 Possible role of Hippo pathway component activation in AKl
In mammalian cells, the Hippo pathway is regulated by a variety of intrinsic and extrinsic factors. Cell polarity, cell-cell junctions, soluble factors, mechanical cues, and metabolic states, such as cellular energy and oxygen stress, can modulate Hippo pathway activity (Boggiano & Fehon, 2012;Meng, 2016; Mo et al., 2015; Zhao et al., 2011b). In particular,mechanical cues, such as the ECM and cellular geometry, are potent regulators (Dupont et al., 2011). For example,YAP/TAZ can be activated when cells are cultured on a matrix with high stiffness or large surface area. In addition, high density of round and compact cells can lead to YAP/TAZ activation. Conversely, low density of flat and loose cells can result in Hippo pathway inactivation, allowing YAP/TAZ to enter the nucleus and promote transcription of target genes(Aragona et al., 2013; Wada et al., 2011). Both apical and basolateral spectrin networks play regulatory roles in the Hippo pathway and function as sensors of mechanical cues(Deng et al., 2015; Fletcher et al., 2015).
Soluble factors act primarily through G protein-coupled receptors (GPCRs) in the regulation of the Hippo pathway.GPCRs are the largest family of plasma membrane receptors,linking the Hippo pathway to a broad range of upstream signals. GPCR ligands that signal through Gα12/13 or Gαq/11,such as lysophosphatidic acid (LPA), angiotensin II, and thrombin, increase YAP/TAZ nuclear entry by activating Rho GTPases; whereas ligands that signal through Gαs, such as epinephrine and glucagon, can activate LATS1/2 and repress YAP/TAZ via the AC-cAMP-PKA pathway (Yu et al., 2012,2013; Zhou et al., 2015). Rho GTPases and actin cytoskeleton remodeling are key mediators of GPCR ligands and mechanical cues in the regulation of the Hippo pathway(Figure 2) (Dupont et al., 2011; Zhao et al., 2012).
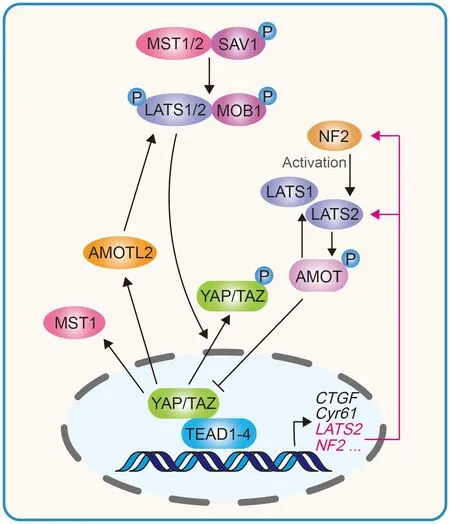
Figure 2 YAP/TAZ induced negative feedback loop in regulating Hippo pathway homeostasis
In addition to GPCR ligands, the Hippo pathway also participates in crosstalk with Wingless/Ints (Wnt) (Bernascone& Martin-Belmonte, 2013), transforming growth factor (TGF)-β(Feng et al., 2018b; Seo et al., 2016), bone morphogenetic proteins (BMPs), Notch, Hedgehog (Hh) (Hansen et al., 2015),Src (Kim et al., 2019), Rac-GTPase activating protein 1(RacGAP1) (Zhou et al., 2020), Gsk3β-p53 (Li et al., 2019),and EGF receptor (EGFR)-phosphatidylinositol 3-kinase(Pl3K)-Akt (Chen et al., 2018), which modulate YAP/TAZ activities and collectively control tissue growth and development. The EGFR-PI3K-Akt pathway and RacGAP1 can help renal tubular epithelial cells (RTECs) recover from acute injury by activating YAP (Chen et al., 2018; Zhou et al.,2020), whereas Wnt5a can exacerbate TGF-β1-induced macrophage M2 polarization and renal fibrosis through YAP/TAZ up-regulation (Feng et al., 2018b). These Hippo pathway regulatory factors may provide potential drug targets for the treatment of kidney injury.
NEGATlVE FEEDBACK LOOP OF HlPPO PATHWAY
Long-term hyperactivation of YAP/TAZ can lead to massive tissue overgrowth, enhanced stem cell-like properties that promote tumorigenic potential, and increased metastasis by epithelial-mesenchymal transition (EMT) (Harvey et al., 2013;Lei et al., 2008). Therefore, YAP/TAZ activity must be tightly controlled to maintain tissue homeostasis. In mammalian cells,the Hippo pathway exhibits a negative feedback mechanism.YAP/TAZ and TEAD complexes directly induce the transcription of NF2 and LATS2, and indirectly stimulate LATS1/2 kinase activity by increasing NF2 protein abundance.A reciprocal negative regulation also exists between YAP and TAZ dependent on LATS and proteasomal degradation(Moroishi et al., 2015; Park et al., 2016). YAP/TAZ-induced LATS activation can lead to increased angiomotin (AMOT)phosphorylation and protein accumulation. AMOT can directly inhibit YAP/TAZ and indirectly activate LATS, thus contributing to the negative feedback loop (Zhao et al., 2011a).Furthermore, MST1 and AMOT-like 2 (AMOTL2) are upregulated by YAP/TAZ, and AMOTL2 can also activate LATS(Kim et al., 2016; Paramasivam et al., 2011; Park et al., 2016).This negative feedback loop is essential for maintaining proper transient activation of YAP/TAZ in response to stimuli and preventing tumorigenesis. Aberrant increase of OGlcNAcylation on LATS2 can disrupt the LATS-mediated negative feedback loop, leading to YAP/TAZ hyperactivation and carcinogenesis (Kim et al., 2020).
RENAL DYSFUNCTlON CAN BE EXACERBATED BY HlPPO PATHWAY-MEDlATED MlTOCHONDRlAL FlSSlON FOLLOWlNG AKl
The kidney is second only to the heart in mitochondrial count and oxygen consumption (Duann & Lin, 2017). Mitochondria undergo significant changes after AKI, which affects the pathophysiology and recovery of renal function (Baligand et al., 2017; Funk & Schnellmann, 2012; Lan et al., 2016).Persistent disruption of mitochondrial homeostasis and sustained tubular damage can result in progression to CKD(Jiang et al., 2020). The pathogenic mechanisms of mitochondrial dysfunction include mitochondrial DNA damage(Lee & Back, 2017), mitophagy disturbance (Duann et al.,2016), and biosynthesis disorder (Xiao et al., 2017). RTECs are rich in mitochondria and the proximal tubule is especially vulnerable to mitochondrial damage (Hall et al., 2009).Mitochondrial apoptosis is considered the main reason for reperfusion-mediated RTEC apoptosis under renal ischemia/reperfusion (I/R) injury, attributable to mitochondrial fission (Perry et al., 2018; Wang et al., 2020b).
The core proteins that mediate mitochondrial fission and fusion include dynamin-related protein 1 (Drp1), mitofusin(Mfn), and optic atrophy 1 (OPA1). Drp1 mediates mitochondrial fission, whereas Mfn and OPA1 mediate fusion of the outer and inner membranes, respectively (Lee & Yoon,2016). Li et al. (2019) demonstrated that MST1 is markedly up-regulated after renal I/R injury and that higher MST1 expression is associated with Gsk3β-p53 axis activation,which induces mitochondrial fission by Drp1 phosphorylation and F-actin assembly. Mitochondrial fission contributes to mitochondrial potential reduction, reactive oxygen species(ROS) production, mitochondrial permeability transition pore(mPTP) opening, cytochromec(cytc) release, and caspase-9-related mitochondrial apoptosis initiation, leading to RTEC apoptosis and renal dysfunction. Feng et al. (2018a) also showed that MST1 expression is up-regulated in response to renal I/R injury and that genetic ablation of MST1 improves renal function, alleviates reperfusion-mediated RTEC apoptosis, and attenuates kidney vulnerability to I/R injury.Moreover, increased MST1 blocks the AMPK-YAP pathway,thereby diminishing OPA1 expression, repressing mitophagy,and activating mitochondrial apoptosis in reperfused kidneys.Reactivation of the AMPK-YAP-OPA1 signaling pathway provides a survival advantage for RTECs in the context of renal I/R injury by repressing mitochondrial fission (Figure 3).Based on these findings, we speculate that the Hippo pathway plays a vital role in the pathogenesis of AKI and that increased MST1 expression can mediate mitochondrial damage and RTEC apoptosis by modulating mitochondrial fission.
YAP OVEREXPRESSlON lN RTECs CAN RESULT lN ACQUlRED CYTORESlSTANCE AFTER AKl
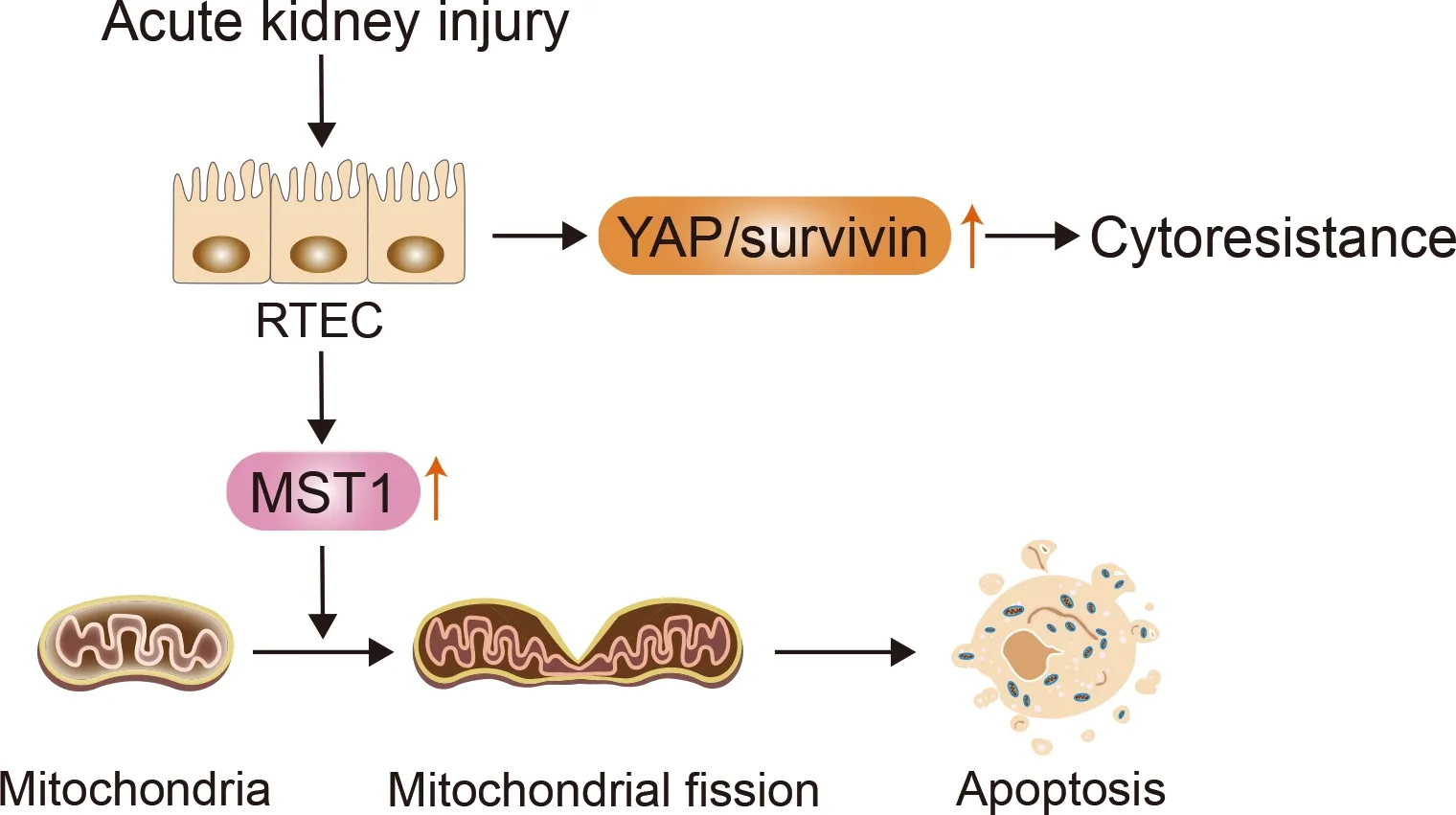
Figure 3 lncrease in YAP and MST1 following AKl can result in RTEC cytoresistance and apoptosis, respectively
Animals recovering from previous AKI are resistant to subsequent renal challenge with the same agent, termed acquired resistance (Honda et al., 1987). The emergence of acquired cytoresistance after AKI can serve to protect the kidneys from further ischemia or nephrotoxicity (Zager, 2013).This is closely associated with heat shock proteins (Furuya et al., 1997; Mizuno et al., 1997), hypoxia-inducible factor (HIF)-1α (Ma et al., 2009), heme oxygenase-1 (Nath, 2014), cyclindependent kinase inhibitor (CDKI) (Nishioka et al., 2014; Price et al., 2004), growth/proliferation factors (Sun et al., 2011),and DNA repair-related factors (Miyaji et al., 2001; Sano et al.,2000).
Iwakura et al. (2017) demonstrated that rat renal proximal tubule epithelial cells (RPTCs) that have recovered from uranyl acetate (UA)-induced AKI can develop cytoresistance to subsequent UA treatment. These cells transiently overexpress YAP and its downstream mediatorsurvivin, which may inhibit apoptosis and increase acquired cytoresistance.These effects may be correlated with enhanced G1 arrest via modulation of cell-cycle regulating factors, including cyclin D1,p21, and p27 (Iwakura et al., 2016), and persist until YAP/survivinexpression levels return to baseline (Figure 3).Furthermore, YAP overexpression leads to hyperplasia in RPTCs, while YAP regression leads to a gradual decline in RPTCs with enhanced cell cycle arrest. Based on these findings, considerable attention should be given to the relationship between RTEC cytoresistance and cellular recovery with YAP/survivinexpression in AKI treatment.
TRANSlENT YAP/TAZ ACTlVATlON PROMOTES RTEC ADAPTlVE REPAlR lN MlLD/MODERATE AKl
Due to the high abundance of mitochondria, the metabolically active proximal tubule, especially the S3 segment, is a primary target of injury in AKI (Munshi et al., 2011). Renal stem cells can be a cell source for maintaining normal kidney cell turnover (Alexandre et al., 2009; Bruno et al., 2009; Morigi et al., 2008). Mouse kidney progenitor cells (MKPCs) are a unique renal stem cell population, which can contribute to renal repair and survival after ischemic injury (Lee et al.,2010). Notably, intrarenal injection of MKPCs isolated from the medullary and papillary interstitium in ischemic-injured mice can rescue renal damage, with some MKPCs forming vessels with red blood cells inside and some incorporating into renal tubules 7 days after injury. Intrinsic RTEC dedifferentiation,proliferation, and migration can also contribute to renal regeneration after AKI (Duffield & Humphreys, 2011; Lin,2006). Surviving RTECs can regenerate and regain normal kidney function if the injury is mild and transient, but may exhibit maladaptive repair if the injury is severe and recurring,leading to renal fibrosis (Figure 4) (Ferenbach & Bonventre,2015; Qi & Yang, 2018; Yang et al., 2011). Recent studies have demonstrated the pivotal role of the Hippo signaling pathway in RTEC recovery and regeneration after AKI.
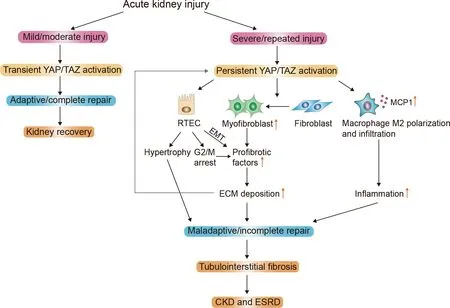
Figure 4 Schematic of Hippo pathway mediating renal repair after AKl
Chen et al. (2018) reported enhanced YAP expression and nuclear distribution in the RPTCs of kidneys in patients with AKI and mice with I/R AKI. Furthermore, YAP antagonist verteporfin (VP)-treated and RPTC-specific YAP knockout mice both showed more severe kidney injury compared with the controls, indicating that EGFR-Pl3k-Akt signaling mediates YAP activation and subsequent target genecyclin Dandamphiregulin(AREG) expression.Cyclin Dcan induce the phosphorylation of the retinoblastoma protein (Rb), thus activating cyclin-dependent kinase (CDK) 4 and 6, which are important for G1 to S transition during cell proliferation(Hamilton & Infante, 2016; Musgrove et al., 2011). In addition,AREGacts as an EGFR ligand to further initiate EGFR-Aktdependent YAP activation, thereby promoting adaptive repair of the kidney after AKI. Of note, Chen et al. (2018) also suggested that YAP, but not TAZ, is critical for renal recovery from I/R injury as histological recovery is delayed in the YAP/TAZ double knockout mice but not in the TAZ single knockout mice. In contrast, Wu et al. (2020) reported a beneficial role of tubular TAZ in ischemic AKI, inconsistent with the findings of Chen et al. (2018). They demonstrated thatin vivotreatment with chloroquine, which can enhance TAZ expression in RPTCs, preserves renal function, ameliorates tubular injury, and reduces apoptosis in the renal cortex after I/R injury. In contrast, both TAZ antagonist VP treatment and TAZ knockdown exaggerate tubular apoptosis and renal failure after I/R injury. Further studies are essential to clarify the role of TAZ in RTECs after AKI and illustrate the potential therapeutic targets against AKI.
Transiently enhanced and evenly distributed YAP levels in the cytoplasm and nucleus are reported in mild I/R AKI rat models (Xu et al., 2016). Dedifferentiation markers, including vimentin, α-smooth muscle actin (α-SMA), and proliferatingcell nuclear antigen (PCNA), show similar expression patterns to YAP during recovery after AKI, whereas quiescent tubular markers, including Na+/K+-ATPase and aquaporin 1 (AQP1),are decreased or absent, indicating that YAP may promote the proliferation and redifferentiation of reconstituted epithelia after acute I/R AKI. Moreover, suppression of YAP1 by the N6-methyladenosine mRNA methylase METTL4 promotes renal I/R injury progression (Xu et al., 2020b). Inhibition of YAP/TAZ degradation and nuclear factor-κB (NF-κB)translocation through Rictor/mTORC2 signaling is crucial for preventing renal inflammation and lipopolysaccharide (LPS)-induced AKI, thus revealing the potent effects of YAP/TAZ on alleviation of AKI severity (Figure 4) (Gui et al., 2020).
PERSlSTENT YAP/TAZ ACTlVATlON lN SEVERE AKl LEADS TO MALADAPTlVE REPAlR AND CKD
The Hippo pathway may exhibit bidirectional functions during I/R AKI repair. Xu et al. (2016) demonstrated that YAP mainly elicits beneficial effects on the proliferation of injured RTECs during repair, but persistent activation of YAP can impede the redifferentiation of dedifferentiated tubular cells and promote kidney fibrosis. Notably, they found more pronounced YAP activation in rats four weeks after I/R injury in the incomplete repair (I/R 45 min) group than in the complete repair (I/R 30 min) group. Furthermore, connective tissue growth factor(CTGF), fibrotic protein mRNA (including type I, III, and IV collagen), and RTPCs in the G2/M-phase were up-regulated with YAP increase, indicating kidney fibrosis exacerbation. In addition, the incomplete repair (I/R 45 min) group treated with YAP agonist digitoxin showed more severe tubulointerstitial fibrosis (TIF) compared with the control group. Xu et al. (2021)also implicated YAP activation in the post-acute phase of AKI in renal dysfunction and TIF, although it elicits a beneficial effect in the acute phase. Transcription factor KLF4 can cause persistent activation of YAP, while inhibition of the KLF4-YAP pathway can attenuate IR-induced renal function deterioration and slow TIF progress by decreasing the expression levels of TGF-β and CTGF. Zheng et al. (2021) confirmed that severe ischemic AKI can induce tubular maladaptive repair and TIF by persistently activating YAP. YAP activation can further cause monocyte chemoattractant protein 1 (MCP-1)overexpression and promote renal macrophage inflammation,a driving force for progressive TIF and AKI-CKD transition.
Moreover, down-regulation of YAP can significantly suppress chronic renal fibrosis. Bothin vivoandin vitrostudies report that miR-101a can potentially slow chronic renal fibrosis by blockade of the YAP-TGF-β-Smad signaling pathway via KDM3A (Ding et al., 2020). Kindlin-2 depletion reduces MOB1 degradation and alleviates renal fibrosis via activation of Hippo/YAP signaling in Kindlin-2 knockout mice with unilateral ureteral obstruction (UUO) (Song et al., 2019).Most recently, knockdown of YTHDF1, a modulator of m6A methylation, was shown to alleviate progression of renal fibrosis in both cultured cells induced by TGF-β administration and UUO model mice via down-regulation of YAP (Xing et al.,2022). In conclusion, appropriate modulation of the Hippo signaling pathway, especially intervention in YAP overexpression during repair, may be a potent preventive target in AKI-CKD transition after I/R injury.
Kidney fibrosis can be induced through Hippo pathwaymediated EMT
The severity and frequency of RPTC injury can determine whether the repair mechanism leads to complete recovery or maladaptive repair (Takaori et al., 2016). Maladaptive repair,i.e., renal fibrosis, is characterized by cell cycle arrest at the G2/M phase and a senescence-associated secretory phenotype (SASP). RTECs may turn to a mesenchymal phenotype via EMT after injury, which is a potential source of profibrotic cytokines and ECM deposition, thus leading to gradual aggravation of renal fibrosis (Qi & Yang, 2018).
EMT is the process by which a polarized epithelial cell adopts a mesenchymal cell phenotype, and includes enhanced migratory ability, invasiveness, resistance to apoptosis, and secretion of ECM (Kalluri & Weinberg, 2009).EMT is manifested by a decrease in epithelial markers, such as E-cadherin and zonula occludens-1 (ZO-1), and the acquisition of mesenchymal proteins, including vimentin, α-SMA, fibronectin, and collagen I (Liu, 2010). Anorga et al.(2018) reported that renal injury can result in elevated TGF-β1 expression, leading to TAZ protein induction and nuclear accumulation. Stable TAZ overexpression in RTECs promotes fibrotic factor expression (including CTGF, fibronectin, and plasminogen activator inhibitor (PAI)-1)), dedifferentiation (as evident by altered morphology, increased mesenchymal marker expression, and loss of epithelial characteristics), and growth inhibition via Smad3-dependent CTGF up-regulation.Seo et al. (2016) found that TEC-specific deletion of Sav1 can increase TAZ protein expression and YAP1 nuclear localization after UUO, resulting in TGF-β and TGF-β receptor II up-regulation and aberrant activation of Wnt/β-catenin signaling, thereby aggravating renal TIF and enhancing EMTlike phenotype changes. Sav1 loss can also induce Stat3 activation and SASP with the accumulation of collagen and vimentin, leading to a fibrosis phenotype that can be further enhanced by AKI with aristolochic acid (AA) (Leung et al.,2017). Treatment with VP (YAP inhibitor) inhibits the activation of genes associated with senescence, SASP, and Stat3, and impedes the development of fibrosis (Leung et al., 2017).Therefore, inhibition of the Hippo pathway may mediate the occurrence of EMT, and YAP/TAZ may serve as a new therapeutic target for chronic renal fibrosis progress after AKI.
Crosstalk between Hippo pathway and fibroblasts/myofibroblasts
Myofibroblasts are important in ECM production during TIF.Myofibroblasts are rare in normal kidneys but markedly increase during chronic fibrosis, with possible precursors including interstitial stromal cells, such as fibroblasts,pericytes, and mesenchymal stem cells (MSC), as well as bone marrow-derived cells and endothelial cells. Current evidence suggests that stromal cells are the major origin of myofibroblasts (Iwano et al., 2002; LeBleu et al., 2013; Yuan et al., 2019). Whether epithelial cells can turn into myofibroblasts has been debated for many years. However,recent studies suggest that epithelial cells contribute little to the myofibroblast population after AKI (Humphreys et al.,2010; Koesters et al., 2010; LeBleu et al., 2013).Myofibroblasts are the major source of ECM proteins, crosslinking enzymes, and inhibitors of matrix-degrading metalloproteinases, characterized by the expression of the classic marker α-SMA (Chen et al., 2020; Gewin, 2018).
Upon insult, maladaptive RTECs cause a switch in the microenvironment of the tubulointerstitial area where myofibroblasts are activated (Qi & Yang, 2018). Resident fibroblasts proliferate and transition into myofibroblasts by profibrotic factors, such as CTGF, which are synthesized and released from injured RPTCs through a YAP activationdependent pathway (Chen et al., 2020). Szeto et al. (2016)suggested that soft ECM inhibits while stiff ECM augments TGF-β-induced Smad2/3 nuclear accumulation mediated by YAP/TAZ, leading to fibroblast-myofibroblast transition and renal fibrosis. Furthermore, treating cells and mice with VP can reduce YAP/TAZ levels and block TGF-β-induced myofibroblast activation. TGF-β1 can also up-regulate YAP/TAZ expression through mTORC2 signaling-stimulated YAP/TAZ transcriptional activation, thereby stimulating fibroblast activation (Gui et al., 2018). Liang et al. (2017)demonstrated that fibroblasts cultured on a stiff matrix transform into myofibroblasts in a process dependent on YAP activation, even in the absence of TGF-β. Furthermore, Gli1+cell-specific knockout of YAP/TAZ in mice suppresses UUOinduced ECM deposition, myofibroblast accumulation, and TIF. Therefore, YAP acts as a tissue mechanosensor in the kidney and can be activated by ECM to transform fibroblasts into myofibroblasts. The interaction between YAP/TAZ and ECM forms a positive feedback loop, resulting in kidney fibrosis.
Correlation between Hippo pathway and G2/M cell cycle arrest in renal fibrosis
Cell cycle arrest in RTECs during maladaptive repair is an important driver of kidney fibrogenesis (Yang et al., 2011). In uninjured kidneys, most RTECs are quiescent and remain in the G0 phase of the cell cycle. Upon injury, cells may enter the cell cycle to replace cells lost during the insult, with some cells arrested in either the G1 or G2 phase to allow time for DNA damage repair (Gewin, 2018). Accumulation of cells arrested in the G2/M growth phase is a common feature of progressive fibrotic kidney disease, resulting in the acquisition of a pathogenic phenotype characterized by sustained synthesis and secretion of profibrotic factors (Bonventre, 2014;Ferenbach & Bonventre, 2015).
As YAP/TAZ are involved in cell proliferation and survival,Yap activation can promote cell cycle progression (Chen et al.,2018; Mizuno et al., 2012). Thus, whether YAP/TAZ are associated with G2/M phase cell cycle arrest warrants investigation. Anorga et al. (2018) found that TAZoverexpressing epithelial cells exhibit epithelial growth inhibition and G2/M cell cycle arrest after injury, consistent with the enhanced expression of growth-arrest genep21,conferring a fibrotic phenotype. Down-regulation of YAP in human laryngeal carcinoma Hep-2 cells induces G2/M cell cycle arrest, reduces G0/G1 phase percentage, and increases apoptosis, indicating that YAP inactivation is related to G2/M arrest (Tang et al., 2019). However, other studies have shown that tubular maladaptive repair in the kidney is associated with YAP activation and that up-regulation of YAP induces an increase in the G2/M- and S-phases and a decrease in the G0/G1-phase in HK-2 cells, thus mediating kidney fibrosis after acute injury (Xu et al., 2016; Zheng et al., 2021). Clearly,these two hypotheses conflict with each other and there may exist a feedback regulation against G2/M phase arrest by triggering YAP activation in the kidney. Furthermore, Gerhardt et al. (2021) suggested the Hippo is involved in early injured RPTCs in mice after moderate AKI based on up-regulation of TEAD1 regulon. They also reported up-regulation of regulators involved in YAP-inactivation and Hippo signaling (e.g.,Sav1,Stk3), as well as effectors of the transcriptional response after Hippo silencing (e.g.,Yap1,Tead 1) and YAP-target genes such asAxlandCtgf. In particular, they identifiedVcam1+/Ccl2+ RPTCs at a late injury stage with marked activation of NF-κB-, TNF-, and AP-1-signaling pathways. This population of RPTCs showed features of SASP but did not exhibit G2/M cell cycle arrest, distinct from other reports of maladaptive RPTCs following kidney injury. However, the exact associations between YAP/TAZ and G2/M cell cycle arrest and whether G2/M cell cycle arrest exists in RPTCs that fail to repair after injury require further investigation.
Function of Hippo pathway in macrophage and inflammation regulation
AKI is typically accompanied by immune activation and inflammation, which involve macrophage infiltration. Recruited macrophages exert versatile effects on kidney repair after injury depending on their polarization form: i.e.,proinflammatory M1 phenotype and reparative M2 phenotype.The predominant M1 phenotype in the early phase of AKI produces proinflammatory cytokines and mediators, which, in turn, exacerbate inflammation (Ferenbach & Bonventre, 2015;Qi & Yang, 2018). The M2 polarization of macrophages in the resolution phase of AKI is beneficial for renal recovery.However, the M2 phenotype may exert a pro-fibrotic influence by promoting TIF, expressing matrix metalloproteases, and secreting pro-fibrotic factors (Gewin et al., 2017). Therefore,persistent activation of M2 macrophages is instrumental in maladaptive repair and fibrosis in CKD (Baek, 2019).
Hippo-YAP pathway activation after ischemic AKI is associated with tubular maladaptive repair and interstitial macrophage infiltration mediated by MCP-1 up-regulation,while inhibition of YAP nuclear translocation by VP attenuates macrophage infiltration and TIF (Zheng et al., 2021). VP treatment in UUO-induced AKI mice also decreases macrophage recruitment and cell adhesion molecule expression (Jin et al., 2020). Tubular cell-specific knockout of MST1/2 promotes the expression of inflammatory factors and macrophage marker F4/80; however, expression returns to physiological levels after YAP deletion in MST1/2 double knockout mice (Xu et al., 2020a). These data suggest that YAP may play a pivotal role in chronic inflammation during maladaptive repair after renal injury.
Feng et al. (2018b) demonstrated that YAP/TAZ induction mediates Wnt5a and TGF-β1-promoted macrophage M2 polarization and contributes to renal fibrosis after UUO or I/R injury in mice, whereas specific ablation of the TAZ gene in macrophages can markedly decrease the M2 phenotype and kidney fibrosis. However, further studies are needed to decipher the exact mechanism by which YAP/TAZ regulate macrophage M2 polarization.
Hippo-YAP pathway activation can lead to renal hypertrophy through RTEC enlargement
Renal hypertrophy is characterized by increased organ size due to cell enlargement rather than cell proliferation (Fine &Norman, 1989). In the kidney, the growth of remaining renal tissue in response to renal tissue loss is known as renal compensatory hypertrophy (RCH) and can help restore normal kidney function following disease or loss of kidney tissue(Sugaya et al., 2000). As the proximal tubule forms the bulk of kidney mass, this nephron segment contributes the most to renal hypertrophy (Hayslett et al., 1968).
In renal epithelial cells, increased nuclear accumulation and transcriptional activity of YAP can trigger activation of the Akt/mTOR signaling pathway and its downstream target S6K1.This leads to silencing of the peripheral tight junction protein zona occludens 2 (ZO-2), which induces an increase in cell size and a higher protein/DNA ratio, thereby triggering RCH(Domínguez-Calderón et al., 2016). ZO-2 is also a positive regulator of the Hippo pathway as it associates with LATS1 and functions as a pathway scaffold (González-González et al., 2021). However, chronic cell expansion beyond optimal conditions may present certain disadvantages, and hemodynamic and metabolic hyperfunction of residual nephrons may prove maladaptive over time (Lafferty &Brenner, 1990; Nath, 1992). Excessive RCH can lead to tubular atrophy, interstitial fibrosis, and progressive decline in renal function (Hostetter, 1995). Therefore, persistent YAP activation after AKI may also cause renal hypertrophy and accelerate progression towards CKD (Figure 4).
POTENTlAL ROLE OF HlPPO PATHWAY lN FERROPTOSlS AFTER AKl
Ferroptosis is characterized by intracellular iron accumulation and lipid peroxidation during cell death (Dixon et al., 2012).The characteristics of ferroptosis differ from necrosis,apoptosis, and autophagy in cell morphology and function (Xie et al., 2016). The morphology of ferroptotic cells includes mitochondrial changes, such as reduced volume, decreased mitochondrial cristae, and increased membrane density(Wang et al., 2021). However, the nucleus remains normal,with no chromatin agglutination (Wang et al., 2020a).Following ferroptosis, intracellular glutathione (GSH)consumption and glutathione peroxidase 4 (GPX4) inactivation occur (Li et al., 2020). Recent studies have shown that ferroptosis participates in various pathological models of AKI and classical ferroptosis inhibitors, such as ferrostatin-1 and liproxstatin-1, are capable of interfering with key molecules in the ferroptosis signaling pathway to resist AKI (Hu et al., 2019;Martin-Sanchez et al., 2017; Skouta et al., 2014; Zilka et al.,2017). GPX4 deletion in mice can spontaneously trigger AKI(Friedmann Angeli et al., 2014), whereas GPX4 up-regulation prevents AKI (Zhang et al., 2021).
A growing number of studies have revealed the relationship between the Hippo pathway and ferroptosis in cancer cells.Yang & Chi (2020) suggested that triggering ferroptosis may have therapeutic potential for TAZ-activated tumors.Ferroptosis sensitivity in renal and ovarian cells is regulated by cell density through the TAZ-EMP-NOX4 and TAZANGPTL4-NOX2 pathways, respectively (Yang et al., 2019,2020). TAZ removal confers ferroptosis resistance, while overexpression of the constitutively active form of TAZ(TAZS89A) sensitizes cells to ferroptosis. YAP exerts a similar function in cancer cell ferroptosis, with knockdown of YAP or its direct target gene S-phase kinase-associated protein 2(SKP2) shown to robustly protect cells and abolish lipid peroxidation during erastin-induced ferroptosis (Yang et al.,2021). However, despite current evidence supporting the connection between Hippo pathway effectors and ferroptosis in cancer, there is a lack of research on the role of the Hippo pathway in ferroptosis during AKI progression. Moreover, the specific role of ferroptosis in AKI caused by different etiologies remains unclear. Thus, further exploration in this field is necessary for the clinical treatment of AKI.
HlPPO PATHWAY MAY CONTRlBUTE TO KlDNEY CANCER DEVELOPMENT lN AKl
Renal cell carcinoma (RCC), which is derived from RTECs,accounts for 85% of all renal malignancies (Cinar et al., 2021;Kovacs et al., 1997). Emerging evidence suggests that dysregulation of Hippo-YAP1 signaling plays a significant role in the etiology of aggressive kidney cancer (Han, 2019). YAP1 protein accumulates in the nuclei of clear cell RCC (ccRCC)cells, although normal kidney cells primarily express the cytoplasmic YAP1 protein (Godlewski et al., 2018). Similarly,nuclear YAP1 is considerably higher in ccRCC cells than in unaltered proximal kidney cortices (Rybarczyk et al., 2017).Up-regulation of YAP1 in ccRCC patients is also associated with poorer clinical outcomes (Chen et al., 2014). Thus,nuclear YAP1 appears to play an oncogenic role in ccRCC cells, promoting cell proliferation and survival (Rybarczyk et al., 2017).
Recent studies have identified AKI as a risk factor for renal cancer development (Kusmartsev, 2021). Based on data collected from several multicenter studies, Peired et al. (2020)found that AKI can significantly increase the risk of papillary RCC (pRCC) development and tumor relapse in humans.They also induced I/R injury in wild-type mice and examined their kidneys at different timepoints and found that hypoxia promotes the undifferentiated cell state in various stem and precursor populations by activating Notch-responsive promoters and increasing expression of genes downstream of Notch, driving a hypoxia-inducible factor HIF1a-to-HIF2a switch favoring transformation into cancer stem cells and supporting tumor growth. Notably, YAP/TAZ activity is linked to oxygen availability (Yu et al., 2015). Under hypoxic conditions, HIF1 stimulates the expression of SIAH1/2, two E3 ubiquitin ligases. SIAH1/2 then promote ubiquitination and degradation of LATS2, leading to YAP/TAZ activation (Ma et al., 2015; Xiang et al., 2014). In addition, HIF1 directly induces the transcription of TAZ (Xiang et al., 2014), and YAP interacts with and stabilizes HIF1 to enhance the transcription of HIF1 target genes (Ma et al., 2015). Furthermore, there are two main modalities of YAP/TAZ-Notch signaling crosstalk: i.e.,YAP/TAZ-mediated transcriptional regulation of Notch ligands or receptors and transcriptional co-regulation of common direct target genes by YAP/TAZ and the Notch intracellular domain (NICD) (Totaro et al., 2018). Whether the Hippo pathway exerts a function in pRCC development after AKI deserves further clarification.
Zhou et al. (2021) also investigated the relationship between AKI-associated systemic inflammation and risk of kidney cancer development. They subjected genetically modified mice with Cre-mediated deletion of theTrp53andPtengenes in Ggt1-expressing RPTCs (GPPY mice) to I/R injury to induce acute injury in one kidney. Their results showed that I/R injury-induced AKI promoted the formation of ccRCC in the contralateral uninjured kidney, but not in the kidney subjected to I/R injury. A significant increase in tissue infiltration of neutrophils and fibroblasts was also observed,associated with the up-regulation of inflammatory factors CXCL1, CXCL13, TIMP1, and ILRα, with CXCL1 displaying the highest levels of up-regulation locally and systemically.Notably, CXCL1 expression was predominantly associated with dysplastic epithelial cells in the I/R-injured kidney, but not in the contralateral uninjured kidney. Up-regulation of CXCR2,which serves as a natural receptor for CXCL1, was also observed in stromal fibroblasts, mostly in the uninjured kidney.Administration of anti-CXCR2 antibodies blocked CXCL1/CXCR2 signaling, leading to a reduction in the recruitment of neutrophils, fibroblasts, and most importantly, RCC incidence.Chen et al. (2018) also demonstrated an increase in YAP expression and YAP-TEAD interaction in RPTCs from post-I/R-injured mouse kidneys. Activation of TEAD1 can lead to an increase in the expression of its target, CXCL1, i.e.,chemokine-mediated neutrophil recruitment (Kusumanchi et al., 2021). Zheng et al. (2021) also proved that renal CXCL2 and CXCR2 are markedly increased in post-AKI mice through tubular YAP activation. Hence, growing evidence suggests that the Hippo pathway is involved in the positive association between ccRCC formation and AKI-induced systemic inflammation.
CONCLUSlONS AND FUTURE PERSPECTlVES
Research published over the last several years has revealed the significant role that the Hippo signaling pathway plays in pathological changes of AKI and subsequent repair. During renal pathological injury in AKI, MST1, the upstream factor of the Hippo pathway, mediates RTEC apoptosis by inducing mitochondrial fission, thereby aggravating tubular damage.Transient overexpression of YAP and its downstream mediatorsurvivinin the aftermath of AKI can result in acquired cytoresistance in RPTCs to protect the kidney against further ischemia or nephrotoxicity. Moreover, the Hippo pathway elicits both beneficial and detrimental effects on recovery after AKI. If the injury is mild/moderate, downstream factors YAP/TAZ can be transiently activated, leading to complete/adaptive renal repair. However, if the injury is severe and recurring, the constant increase and activation of YAP/TAZ can lead to incomplete/maladaptive renal repair and progressive fibrotic CKD via several mechanisms, including EMT, fibroblast to myofibroblast conversion, G2/M phase cell cycle arrest, macrophage infiltration, and renal hypertrophy.Although ferroptosis is implicated in various pathological models of AKI and Hippo pathway effectors are highly associated with ferroptosis in cancer cells, whether the Hippo pathway is involved in ferroptosis in AKI progression requires further investigation. In addition, while AKI is a risk factor of kidney cancer development, including ccRCC and pRCC,whether the Hippo pathway participates in this process remains to be clarified. Hence, despite compelling evidence,the correlation between the Hippo pathway and kidney injury still requires additional research, and the potential mechanisms and signaling pathways need to be further elucidated. Furthermore, pharmacological manipulation of the Hippo pathway for the treatment of AKI is uncertain and requires future exploration.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
C.Z., C.L.L., K.X.X., Z.H.Z., and G.Z.C. conceived and designed this review article. C.Z. wrote the initial draft of the manuscript and drew the figures. J.L. and H.J.W. reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.

- Zoological Research的其它文章
- Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
- Genomics and morphometrics reveal the adaptive evolution of pikas
- Molecular phylogeny and taxonomy of four Remanella species (Protozoa, Ciliophora): A flagship genus of karyorelictean ciliates, with descriptions of two new species