
Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
2022-10-17 03:27ZeXianZhuKazumiMatsubaraFoyezShamsJasonDobryErikWapstraTonyGambleStephenSarreArthurGeorgesJenniferMarshallGravesQiZhouTariqEzaz
Zoological Research 2022年5期
Ze-Xian Zhu, Kazumi Matsubara, Foyez Shams, Jason Dobry, Erik Wapstra, Tony Gamble, Stephen D. Sarre,Arthur Georges, Jennifer A. Marshall Graves,5, Qi Zhou, Tariq Ezaz,*
1 MOE Laboratory of Biosystems Homeostasis and Protection and Zhejiang Provincial Key Laboratory for Cancer Molecular Cell Biology,Life Sciences Institute, Zhejiang University, Hangzhou, Zhejiang 310058, China
2 Institute for Applied Ecology, University of Canberra, Canberra 2601, Australia
3 School of Biological Sciences, University of Tasmania, Hobart, Tasmania 7001, Australia
4 Department of Biological Sciences, Marquette University, Milwaukee, Wisconsin 52322, USA
5 School of Life Science, La Trobe University, Melbourne 3168 Australia
6 Department of Neuroscience and Developmental Biology, University of Vienna, Vienna 1030, Austria
7 Center for Reproductive Medicine, The 2nd Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009,China
ABSTRACT Reptile sex determination is attracting much attention because the great diversity of sex-determination and dosage compensation mechanisms permits us to approach fundamental questions about mechanisms of sex chromosome turnover. Recent studies have made significant progress in better understanding diversity and conservation of reptile sex chromosomes, with however no reptile master sex determination genes identified. Here we describe an integrated genomics and cytogenetics pipeline,combining probes generated from the microdissected sex chromosomes with transcriptome and genome sequencing to explore the sex chromosome diversity in non-model Australian reptiles. We tested our pipeline on a turtle, two species of geckos, and a monitor lizard. Genes identified on sex chromosomes were compared to the chicken genome to identify homologous regions among the four species. We identified candidate sex determining genes within these regions, including conserved vertebrate sex-determining genes pdgfa,pdgfra amh and wt1, and demonstrated their testis or ovary-specific expression. All four species showed gene-by-gene rather than chromosome-wide dosage compensation. Our results imply that reptile sex chromosomes originated by independent acquisition of sex-determining genes on different autosomes, as well as translocations between different ancestral macro- and microchromosomes. We discuss the evolutionary drivers of the slow differentiation and turnover of reptile sex chromosomes.
Keywords: Sex determination; Reptiles;Genomics; Cytogenetics; Sex chromosome turnover
INTRODUCTION
Sex can be determined either by genes on specialized chromosomes (genotypic sex determination, GSD) or by environmental factors (environmental sex determination,ESD). Much of our knowledge on sex chromosome evolution has come from studies of model organisms such asDrosophila, chicken and mammals (principally humans and mice), in which species master sex determining genes have been identified (Bachtrog, 2013). Their heteromorphic sex chromosomes can be easily identified by cytogenetic observations because the male-specific Y chromosome, or the female-specific W chromosome is morphologically different from the X or Z chromosome. Sex chromosome differentiation occurs as the result of suppression of recombination, and is manifested by massive accumulation of transposable elements and inactivation or loss of genes (Charlesworth &Charlesworth, 2000). The sex chromosomes of many model vertebrate species have been evolutionarily stable for more than 100 million years, judging from the homology of the pair of sex chromosomes within their clade (Cortez et al., 2014;Zhou et al., 2014).
Like birds and mammals, some reptiles, amphibians and fish have stable sex chromosomes without frequent transitions(Augstenová et al., 2021; Bellott & Page, 2021; Cornejo-Páramo et al., 2020; Giovannotti et al., 2017; Kostmann et al.,2021; Kratochvíl et al., 2021b; Mezzasalma et al., 2021; Rupp et al., 2017; Stöck et al., 2021; Thépot, 2021). However, for majority of reptiles, amphibians and fishes, frequent transitions between different sex determination mechanisms have been observed (Ezaz et al., 2009b; Mank & Avise, 2009; Miura,2017). Reptiles represent an extraordinary variety of sex determining mechanisms, including genotypic sex determination (GSD) XY and ZW systems with varying degrees of sex chromosome differentiation and temperature dependent sex determination (TSD) (Alam et al., 2018). In recent years evolution and transitions of reptiles sex chromosomes and sex determination have received renewed attention due to advances in genome sequencing and integration of cytogenetics knowledge, which revealed novel insights into the dynamic evolution and novel mechanisms in multiple reptile lineages including squamates and chelid turtles, and have been highlighted in multiple recent reviews(Bellott & Page, 2021; Kratochvíl et al., 2021b; Mezzasalma et al., 2021; Rupp et al., 2017; Thépot, 2021). However, genomic landscape of sex chromosomes and sex determining genes in many other reptiles remained uncharacterised.
The evolutionary variety of vertebrate sex determining systems has long been recognized. Cytological observations and limited gene mapping data revealed that multiple transitions between ESD and GSD, and between XY and ZW sex chromosome systems, had occurred in reptiles (Ezaz et al., 2009b), teleost fish (Mank & Avise, 2009) and anurans(Miura, 2017). However, despite this variety, extensive cytogenetic mapping of the reptile orthologues of genes that are located on sex chromosomes of model organisms (e.g.,human and chicken) revealed a surprisingly frequent overrepresentation of particular ancestral autosomes or genomic regions (Deakin & Ezaz, 2014; Ezaz et al., 2017; O'Meally et al., 2012). However, testing the hypothesis on the existence of a conserved ancestry of amniote sex chromosomes will require in-depth comparative analysis on much wider phylogenetic context (Kratochvíl et al., 2021a).
With the development of long-read sequencing and Hi-C technologies, many genomic consortia (e.g., Vertebrate Genome Project (Rhie et al., 2021) and the Earth Biogenome Project (Lewin et al., 2018)) aim to finish the complete genomes of most vertebrate species in the next few years.However sequencing projects usually represent sex chromosomes poorly; either the homogametic sex (XX female or ZZ male) is sequenced, with the male-specific Y or femalespecific W being ignored; or the heterogametic sex only is sequenced with poor representation of the X or Z, and there is great difficulty in assembling the repeat-rich Y or W (Bellott et al., 2017; Skaletsky et al., 2003).
Here, we develop a cost-effective method to identify genes borne on sex chromosomes, combining microdissection of sex chromosomes and high-throughput sequencing, followed by PCR validation and assessment as candidate sex determining genes. A similar method was pioneered to identify novel genes on the Y chromosome of marsupials (Sankovic et al.,2006), and subsequently has been applied to characterise sex chromosome sequences in a plant (Hobza & Vyskot, 2007), in a species of fish (Cocca et al., 2015), two species of anole lizards (Kichigin et al., 2016), in humans (Alvarez-Cubero et al., 2018) and in Gorrila (Tomaszkiewicz et al., 2016). We applied the method to four reptile species, revealing the great diversity of sex chromosomes, and their independent evolutionary origins.
We chose four Australian reptile species, a turtle and three lizard species to represent the variety of reptile sex determining systems (Figure 1). The Murray River turtleEmydura macquarii(Martinez et al., 2008) (referred as “river turtle” hereafter) has minimally differentiated X and Y are macrochromosomes, whereas the pink-tailed worm lizardAprasia parapulchella(Matsubara et al., 2013) (referred as“worm lizard” hereafter) has a highly differentiated XX/XY sex chromosome system in which the X and Y are microchromosomes. The marbled geckoChristinus marmoratus(Matsubara et al., 2014a) (referred as “marbled gecko” hereafter) has a pair of ZZ/ZW sex chromosomes, in which the Z and W heteromorphy involves pericentric inversion, whereas the spiny-tailed monitor lizardVaranus acanthurus(Matsubara et al., 2014b) (referred as “monitor lizard” hereafter) has ZZ/ZW heteromorphic sex chromosomes in which Z and W chromosomes are minimally differentiated microchromosomes (Figure 1).

Figure 1 The diversity of reptile sex chromosomes
MATERIALS AND METHODS
Ethics Statement
Animal care and experimental procedures were performed following the guidelines of the Australian Capital Territory Animal Welfare Act 1992 (Section 40) and conducted under approval of the Committee for Ethics in Animal Experimentation at the University of Canberra (Permit Number: CEAE 11/07 and CEAE 11/12).
Chromosome preparations, sex chromosome microdissection, microdissected chromosome sequencing, probe preparations and FISH analysis
Animal collection, microdissection, preparation of sex chromosome specific probes and validation of probes were described in our previous studies (Matsubara et al., 2013,2014a, 2014b). Briefly, we labelled sex chromosome probes by nick translation incorporating SpectrumGreen-dUTP(Abbott, USA) or SpectrumOrange-dUTP (Abbott, USA) and precipitated with 20 μg glycogen. After decantation, labeled probe pellets were resuspended in a 15 μL hybridization buffer. The resuspended probe mixture was hybridized with a drop of metaphase chromosome suspension fixed on a glass slide, covered with coverslips, and sealed with rubber cement.The slide was then denatured on a hot plate at 68.5 °C for 5 min and was hybridized overnight in a humid chamber at 37°C for two days. The slides were then washed first with 0.4×SSC, 0.3% IGEPAL (Sigma-Aldrich, USA) at 55 °C for 2 min followed by 2×SSC, 0.1% IGEPAL for 1 min at room temperature. The slides were dehydrated by ethanol series and air-dried and then mounted with anti-fade medium Vectashield (Vector Laboratories, USA) containing 20 μg/mL DAPI (4′,6-diamidino-2-phenylindole.). We sequenced single microdisseted sex chromosome from all species except for river turtle (E. macquarii) Y chromosome, where we sequenced a pool of 5 Y chromosomes. We amplified sex chromosome DNA using GenomePlex single cell whole genome Amplification kit (Sigma, USA) following manufacturer’s instruction as described in Matsubara et al.(2013, 2014a, 2014b). Next generation sequencing of the amplified sex chromosomes DNA library was performed at the Beijing Genome Institute (BGI) using Illumina HiSeq2000(USA). We sequenced 500 bp insert library and all reads were trimmed to 90 bp for both ends to get 2 GB clean reads.
Transcriptome assembly and annotation
RNA-Seq data from gonads and brain tissues for males and females of monitor lizard (V. acanthurus), river turtle (E.macquarii) and marbled gecko (C. marmoratus) and tail tissue from a male and female worm lizard (A. parapulchella) (Each tissue has only one sample from one individual due to the difficulty of collecting the tissues from these wild species) were used to performde novoassembly of each species with Trinity v2.4.0 pipeline (Grabherr et al., 2011). Then we used transcoder (Grabherr et al., 2011) to do ORF prediction and cd-hit (v4.7) (Fu et al., 2012) to remove the redundant sequences with the parameters -c 1.00 -b 5 -T 8. For evaluating the quality of the assembly, we examine the number of transcripts that appear to be full-length or nearly full-length by BLAST+ (v2.6.0) (Altschul et al., 1990) with the E-value 1e-3. For worm lizard and marbled gecko,the reference species isGekko japonicuswhile for river turtle,the reference species isPelodiscus sinensis, and transcripts with a minimum 30% coverage of reference were selected.
We used the Trinotate (v2.0.1) (Grabherr et al., 2011)pipeline to annotate the transcriptome. First, we aligned the transcripts to the reference library consisting of human and chicken using blastx and the protein file using blastp with the e-value 1e-3. Also, we used HMMER (v3.1b2) to do another annotation which aligned the transcripts to the Pfam protein library according to the hidden Markov algorithm with the default parameters. Later, the transcripts and the protein,along with the alignments from blast and HMMER were fed to Trinotate to annotate the transcriptome. The transcriptomes were evaluated by assessing the number of fully reconstructed coding transcripts with their reference species, which areG.japonicusfor worm lizard and marbled gecko,andP. sinensisfor river turtle.
Genome assembly and annotation
We used SOAPdenovo2 (v2.0.4) pipeline (Luo et al., 2012) to assemble the Illumina DNA reads from microdissected sex chromosomes. In brief, we first tried several times with default parameters, to find the bestk-mers with the longest N50.Then, we adjusted the average insertion size according to the best result and re-run the scaffold step. Afterwards, we used kgf (v1.16) with the parameters -m 5 -t 6 and Gapcloser(v1.12) to fill the gaps (Luo et al., 2012) , which finally built ade novodraft assembly for sex chromosomes of our species.We constructed the genome assembly of monitor lizard (V.acanthurus) with the Supernova v2.1.1 pipeline (Weisenfeld et al., 2017) with the default parameters, which is a package forde novoassembly based on 10X sequencing. Briefly, the approach is to first build an assembly using readk-mers(default is 48), then resolve this assembly using read pairs (tok=200), then use barcodes to effectively resolve this assembly tok≈100 000. The final step pulls apart homologous chromosomes into phase blocks, which create diploid assemblies of large genomes.
We annotated the genome of monitor lizard (V.acanthurus)with the Braker2 v2.1.5 pipeline (Brůna et al., 2021) which combined evidence of protein homology, transcriptome andde novoprediction. First, we used RepeatMasker (v4.0.7)(Tarailo-Graovac & Chen, 2009) with parameters: -xsmall -species squamata -pa 40 -e ncbi, and the Repbase (v21.01) to annotate the repeat sequences. Then we aligned all available RNA-seq reads to the reference genome by STAR (v2.5)(Dobin et al., 2013) to construct transcriptome evidence. Later we fed the masked genome, the alignment of RNA-seq, and the reference protein sequences, which were human and chicken here, to Braker2 with parameter: --prg=exonerate,setting exonerate for protein homology prediction. Finally, the package outputs the GFF file containing the gene models,along with protein sequences and CDS sequences.Additionally, we also separately annotated the sex chromosome of monitor lizard. First, we aligned our sequences to the reference protein sequences using tblastn with parameters: -F F -p tblastn -e 1e-5. The results were then refined by GeneWise (v2.4.1) (Birney et al., 2004), and for each candidate gene, we kept the one with the best score.Within these genes, we filtered them if premature stop codons or frameshift mutations reported by GeneWise or single-exon genes with a length shorter than 100 bp, or multi-exon genes with a length shorter than 150 bp, or if the repeat content of the CDS sequence is larger than 20% exists.
Sex-linked sequences identification
For worm lizard (A. parapulchella), river turtle(E. macquarii)and marbled gecko (C. marmoratus),using an XY system as an example, we first assembled all the RNA-seq reads into a pooled transcriptome, the female reads into a XX transcriptome, and the male reads into a XY transcriptome.Then male RNA-seq reads were mapped to the XX transcriptome with bowtie2 (v2.2.9) (Langmead & Salzberg,2012) with default parameters. Read depth was then calculated using SAMtools (v1.6) (Li, 2011), and those reads unmapped were assembled into a transcriptome, which was considered to be X reads excluded.
The pooled transcriptomes were directly mapped by Illumina DNA reads from the X and Y chromosomes, and those sequences not mapped by either X or Y reads were assigned as autosomal genes. XX transcriptome and XY transcriptome were both mapped by DNA reads from the X and Y chromosomes, the reads depth (coverage/mappable site) was calculated for each genomic regions mapped, and sequences with a depth higher than 3 and a minimum coverage of 10%with X reads, simultaneously with no alignments with Y reads were assigned as X-linked. For the transcriptome with X reads excluded, the same steps were repeated and sequences with a depth higher than 3 and a minimum coverage of 10% with Y reads and no alignments with X reads were assigned as Ylinked. Afterwards, for sequences with both reads depth (X reads and Y reads) higher than 3, along with a minimum 10%coverage with both X and Y reads, we assigned them as shared genes.
To identify the sex-linked sequences in monitor lizard (V.acanthurus), Illumina reads from both sexes were aligned to the scaffold sequences using bowtie2 with default parameters.Read depth of each sex was then calculated using SAMtools in 10 kb non-overlapping windows and normalized against the median value of depths per single base pair throughout the entire genome for the comparison between sexes. Those sequences with depth ratio of male-vs-female (M/F) ranging from 1.75 to 3, along with a read coverage ratio of male-vsfemale higher than 0.8 were assigned as Z-linked sequences.For the rest of the sequences, those with M/F ratio of depth and coverage both ranging from 0.0 to 0.25 are assigned as W-linked sequences, and the remaining are assigned as autosomes. Those annotated genes in the sex-linked sequences were assigned as Z-borne or W-borne genes.Later we aligned the Z-borne genes to the W-borne genes with blastn, and those genes that have an identity higher than 0.8 and aligned length >500 bp were assigned as shared genes.
Homology comparisons
To find the orthologs of our genes with chicken, we compared the sex-linked transcripts of worm lizard (A. parapulchella),river turtle (E. macquarii) and marbled gecko (C. marmoratus),and the sex-linked genes annotated of the monitor lizard (V.acanthurus) 10× assembly to the proteins of chicken (v6,Ensembl), respectively using blastx with the e-value 1e-5. The result was filtered with the aligned AAs >30% coverage of the reference chicken protein, along with a minimum 50% identity,and returned the one-to-one best hits, with the duplications retained. Then we merged the alignment sites from the four species and calculated the total number of orthologs on the relative chicken chromosomes. With the same protocols, we found the orthologs of our sex-linked genes with chicken sexdetermining genes, except for the threshold of identity which were adjusted to 40%.
Gene expression analyses
To quantify gene expression, RNA-seq reads were mapped to the transcripts of worm lizard (A. parapulchella),river turtle (E.macquarii) and marbled gecko (C. marmoratus) and the CDS of monitor lizard (V. acanthurus) by bowtie2. The raw read counts were estimated by RSEM (v1.3.1) (Li & Dewey, 2011),with both TPM expressions calculated. Those genes which have orthologs with chicken were filtered for dosage compensation analysis. Correlation within sexes for each species was tested using a Wilcoxon rank-sum test, where a significant differentiation within samples was found, with aPvalue smaller than 0.05.
Gonadal biased genes were identified by calculating the fold change of gonadal to somatic expressions of the four species.For both ovary and testis, bias genes were classified into 4 categories of TPM ratio, namely <2, 2 to 3, 3 to 5, >5. For those genes that have a ratio <2 are assigned as negative, for those ≥2, are assigned as gonadal biased, and only those genes with a ratio higher than 5 were calculated in the correlation test for masculinization.
dN/dScalculation
To calculate thedN/dSvalues between the gametolog genes of monitor lizard and Komodo dragon, proteins sequences of Z or W gametologs from monitor lizard were aligned to the proteins of Komodo dragon, and the reciprocal-best-hit of proteins were identified by blastp with the identity level higher than 70 and the aligned amino acids higher than 70% of the protein length. Then, we aligned the coding sequence of each pair of genes using Prank (v.150803) with default parameters,anddN/dSwere later calculated for each pairwise alignment using the codeml module of PAML (v4.8) (runmode=2).
INDEL calling and PCR validation of sex specific markers
Using the identified 10.81 Mb Z-borne scaffolds and 7.10 Mb W-borne scaffolds of monitor lizard (V. acanthurus),we first identified indels based on their alignment using LASTZ(v1.02)(Harris, 2007) with default parameters. On two homologous scaffolds (ChrZ_scaf_189 and Chr_W_scaf_176),we found three W-specific insertions with the lengths as 206 bp, 331 bp and 209 bp (Supplementary Table 7). At the flanking regions of these insertions, we designed two PCR primers spanning a predicted length of 1 312 bp (assay 1) and 2 479 bp (assay 2) sequence fragments for validating the sex chromosome specificity in monitor lizard (V. acanthurus). Both primer sets were amplified under the following conditions:initial denaturation at 95 °C for 5 min followed by 35 cycles of 95 °C for 30 s, 57 °C for 30 s and an extension step of 72 °C for 1 min, with final extension of 72 °C for 10 min. These two markers were validated on 60 individuals; 22 females and 38 males from five different localities distributed across the species distribution.
RESULTS
Transcriptome and genome assemblies of sex chromosomes of the four reptile species
The four reptile species have cytologically distinguishable sex chromosome pairs (Figure 1; Supplementary Figure S1);these were morphologically differentiated in the worm lizard and the monitor lizard, but more subtle for the river turtle and the marbled gecko (Matsubara et al., 2013, 2014a, 2014b).For each species, we microdissected each of their sex chromosomes, performed linear genome amplification and validated the sex chromosome specificity of the DNA products by chromosome painting (Figure 2A; Supplementary Figure S1).
For each sex chromosome, we generated up to 2 Gb clean paired-end (PE) Illumina reads from the microdissected sex chromosome DNA (Supplementary Table S1). To identify genes borne on sex chromosomes, we also produced 2 Gb transcriptomes per sample from the gonad and brain tissues for males and females of monitor lizard, river turtle and marbled gecko and somatic transcriptomes (tail tissue) from a male and female worm lizard (Figure 2B). Genomic reads derived from each sex chromosome were then used to identify sex-linked genes fromde novoassembled transcript sequences of each species. We annotated a total of 11299,15202 and 10507 non-redundant transcripts, respectively for the worm lizard, the marbled gecko, and the river turtle,using chicken genes as a reference for each.
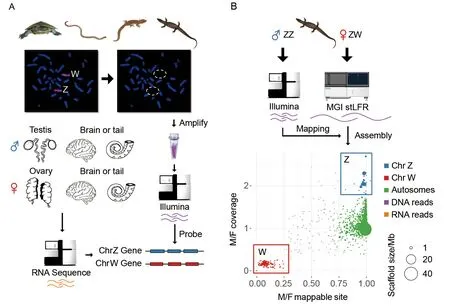
Figure 2 Transcriptome and genome assemblies of four Australian reptile species
For the monitor lizard, inferences based on transcripts and microdissected sex chromosome sequences were uncertain because its sex chromosomes were reported to have originated from translocations between fragments of multiple ancestral autosomes (also see below) (Iannucci et al., 2019),as well as the poor quality of sequences obtained from microdissected monitor lizard sex chromosomes. Therefore,we further generated 200 Gb (135× genomic coverage) singletube long fragment linked reads (stLFR)(Wang et al., 2019)from a female individual, and 30 Gb Illumina PE reads from a male individual, so that we can identify the sex chromosomes by comparing reads between sexes. We performedde novogenome assembly and produced a female draft genome with the total length of 1.46 Gb and the scaffold N50 length of 12.8 Mb (Supplementary Table S2). The high continuity of the draft genome was evident from 94 very large scaffolds that accounted for 80% of the entire genome. Using protein sequences of human and chicken as reference, we annotated a total of 14 521 genes for the monitor lizard and identified its sex-linked sequences based on the comparisons of mapped read patterns between sexes. The putative W-linked scaffolds showed female specificity in both their mapped read number and mapped sites, whereas the Z-linked scaffolds showed a 2-fold increase of male mapped reads compared to that of female mapped reads, but an equal number of mapped sites for putative autosomal scaffolds between sexes (Figure 2B).Using this approach, we identified 10.81 Mb Z-linked scaffolds with 333 genes, and 7.10 Mb W-linked scaffolds with 87 genes.
Identification of sex-linked genes
To identify the sex chromosome-borne genes in three species other than the monitor lizard, we developed a pipeline to separately assemble transcripts of genes that are X- or Yborne (or Z- and W-borne) using the sexed transcriptomes(Figure 3A). In brief, we considered that transcripts that were assembled using pooled RNA-seq reads of both sexes and could not be aligned using the sex chromosome DNA probes were autosomal genes. Conversely, male RNA-seq reads in XY species that could not be aligned to the female transcripts were assembled into candidate Y- borne transcript sequences.Then by comparing the mapped read numbers of Y- or Xborne probes for each candidate Y-borne transcript or each transcript assembled from female RNA-seq, we were able to categorize them into the genes that were specific to the X or to Y chromosome, or were shared between X and Y. We also conducted the same process for the ZW marbled gecko but in reverse. Those shared genes however cannot be assigned as either X/Z- or Y/W-linked.

Figure 3 Processes involved in the identification of sex-linked genes
Following our stringent filtering criteria (see Methods), we identified 193 X-borne or X-linked hemizygous genes, 1 Yborne gene and 17 shared genes between X/Y chromosomes in the worm lizard,21 X-borne genes, 7 Y-borne genes and 218 shared genes in the river turtle and 50 Z-borne genes 37 W-borne genes and 307 shared genes in the gecko,281 Zborne genes 35 W-borne genes and 52 shared genes in the monitor lizard (Figure 3B; Supplementary Table S3).We considered these numbers to be conservative estimates of the sex chromosome-borne genes in these species because genes with low expression levels may not be well assembled in our transcriptome data, and there could also be a sampling bias in the sex chromosome probes captured by microdissection. We further designed primers spanning regions of insertions or deletions between the sex chromosomes and confirmed their length variations between sexes by PCR for the sex chromosome-borne genes of the monitor lizard (Figure 3C). We found no indel sequences within the coding regions of sex chromosome borne genes of the other three species, hence did not design primers for validation.
The proportion of genes that are specific for one or other sex chromosome, and the proportion that are shared, provide a good indication of the degree of genetic differentiation of the sex chromosome pair, and correlate well with our cytogenetic observations (Supplementary Figure S1). The high numbers of genes shared between the X and Y chromosomes of the river turtle suggest that its sex chromosome pair is not highly differentiated, which is consistent with the subtle difference in size and morphology of the X and Y (Figure 1). Among the three lizard species, the numbers of shared versus sex chromosome-specific genes also implied different degrees of sex chromosome differentiation. Consistent with the cytogenetic data (Figure 1; Supplementary Figure S1)(Matsubara et al., 2013), the Z and W chromosomes of the marbled gecko also shared most genes, whereas the monitor lizard showed an intermediate level of shared Z- and W-borne genes. In contrast, the worm lizard had many X-specific but very few Y-specific genes, and only a few X-Y shared genes,implying that the Y chromosome is highly degraded.
Origins of sex chromosomes of the four reptile species
By mapping the orthologues of sex-linked genes of the four reptiles to the chicken genome (GGA), we found evidence for both independent origin and convergent evolution of sex chromosomes (Figure 4; Supplementary Figure S2).
In each species, genes borne on the sex chromosomes clustered together predominantly on a single chicken chromosome, though in three of the species there were other minor clusters. Sex chromosomes of the three lizards were homologous to quite different regions of the chicken genome,on chromosomes GGA1, GGA4 and GGA28 respectively,implying independent origins. However, the sex chromosomes of the river turtle largely overlapped with those of the worm lizard on GGA4q, the long arm of chicken chr4. This is unlikely to represent sex chromosome identity by descent, since the turtles are more closely related to birds (in which this region is autosomal) than they are to squamates, with divergence times of ~250 million years ago (Ma) and 285 Ma respectively(Wang et al., 2013), however, in-depth analysis in multiple outgroups is warranted to test this assumption.
Secondary sites of homology between our four reptile species and the chicken genome represent fragments of sex chromosomes with a different evolutionary origin. For instance, homologs of some river turtle sex chromosomeborne genes were found on chicken microchromosome GGA32 (Supplementary Figure 2). This supports the hypothesis that the sex chromosomes of the river turtle originated by a recent translocation between an ancestral sex chromosome pair (GGA4) and a microchromosome pair (Lee et al., 2019b; Martinez et al., 2008).
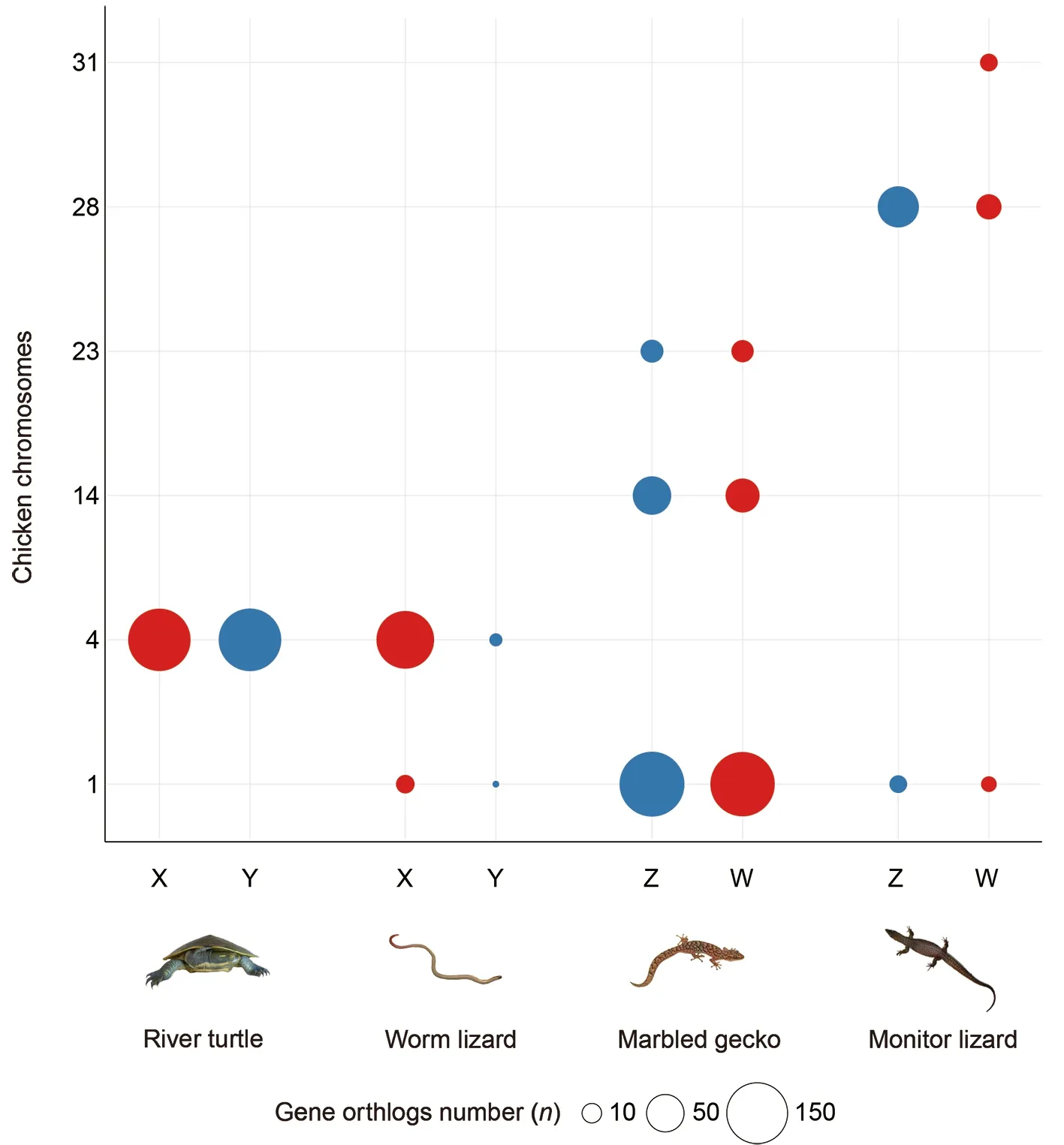
Figure 4 Independent origin of the sex chromosomes of four reptile species
The sex chromosomes of marbled gecko were mainly homologous to GGA1p, with strong secondary signals on GGA14 and GGA23 that contained very similar numbers of Zand W-borne genes. Genes on the sex chromosomes of the monitor lizard were mainly homologous to GGA28, with strong secondary sites at GGA31, GGA33 and Z that contained similar numbers of sex chromosome-borne genes (Figure 4;Supplementary Figure 2).
Candidate sex-determining genes of the four reptile species
Novel sex chromosomes may arise when an autosomal gene acquires a sex determining function or through autosome sex chromosome fusion (Lee et al., 2019a; Pennell et al., 2015).Sex chromosome turnovers have occurred many times during reptile evolution (Bista et al., 2021; Gamble et al., 2015,2017), possibly by a novel sex determining gene usurping the established gene (Herpin & Schartl, 2015) (e.g.,sdYin rainbow trout (Yano et al., 2012), or a change to environmental sex determination and the subsequent evolution of novel genetic systems (Holleley et al., 2015). It would not, therefore, be unexpected to find different candidate sex determining genes within the genomic regions that we have identified in the four reptiles (Figures 1, 4).
To test this, we compiled a list of genes reported to be involved in the sex-determining pathways of other vertebrates(Supplementary Table S4 and Figures S3, S4) and looked for their orthologs among genes that were either identified as sexlinked or fell within the identified sex-linked region in each studied species (Figure 5A, B; Supplementary Tables S5, S6).Included in the region is GGA4q that overlaps with the homologous regions of the river turtle and the worm lizard X chromosomes and contains one candidate male-determining genepdgfra(platelet-derived growth factor receptor alpha).Another candidate sex determining geneAR(Androgen receptor) located on GGA4p was also annotated as X-borne in these two species. This suggests an independent acquisition ofARgene because GGA4p is a microchromosome in all other birds which was fused recently in chicken. Thepdgfa(platelet-derived growth factor alpha polypeptide) gene and its receptorpdgfra(platelet-derived growth factor receptor alpha)have been shown to be critical for testis development,particularly Leydig (male steroidogenic) cell development in mammals and turtles (Brennan et al., 2003; Rhen et al., 2009),whereasARis more likely to be involved in the downstream sexual differentiation process after the gonad sex is determined (Hiort, 2013; Wilson et al., 1980). We confirmedpdgfrato be X-borne in the worm lizard using our transcriptome assembly and sex-linked probes from microdissected sex chromosomes. In the river turtle, we could not annotatepdgfraas a sex chromosome-borne gene because of a lack of mapped sex-chromosome borne probes,but it was embedded among other sex chromosome-borne genes in, so that is likely to be sex chromosome-borne also in the river turtle (Figure 5B).
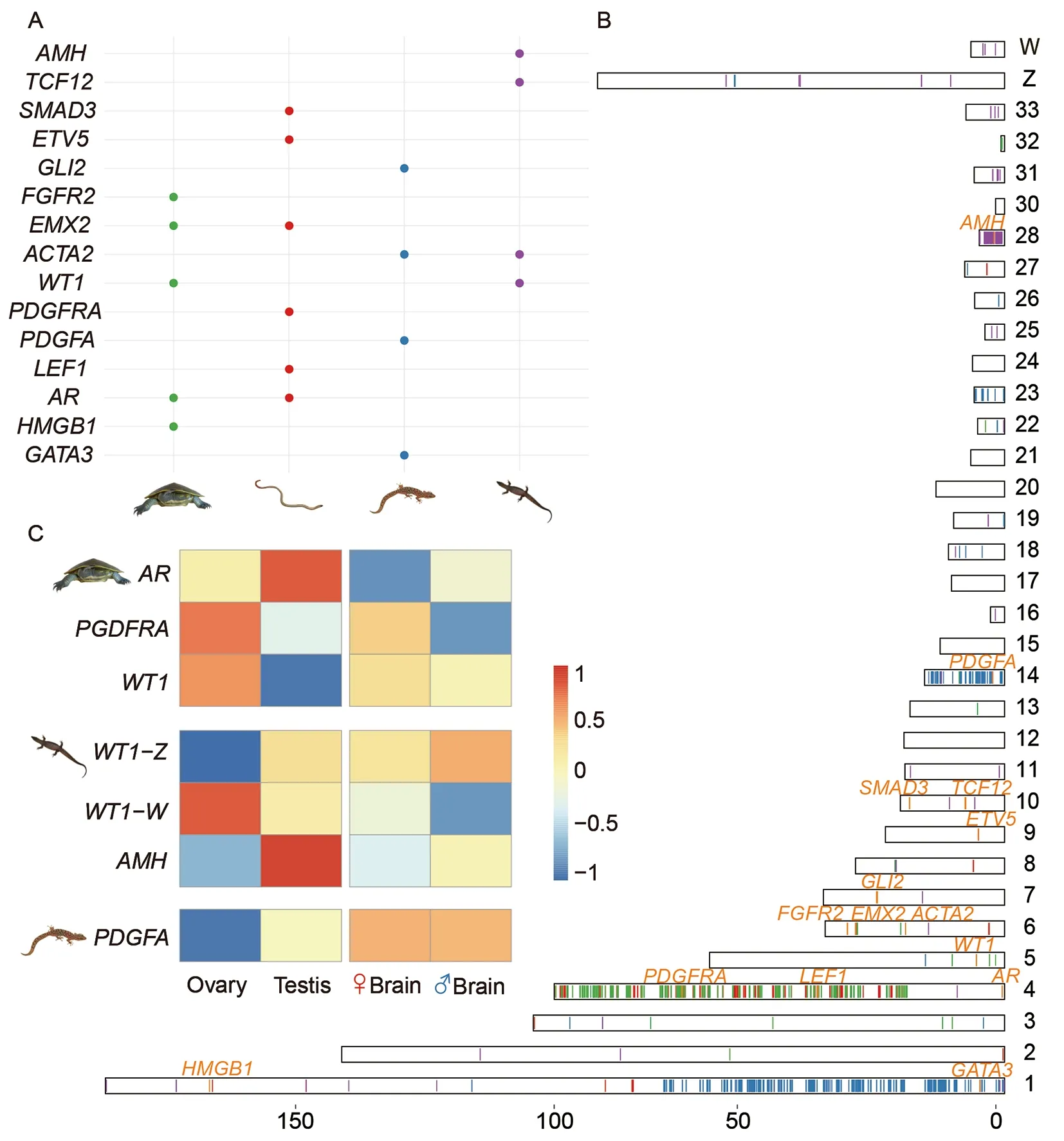
Figure 5 Candidate sex-determining genes of the four reptile species
For the marbled gecko, a promising candidate sexdetermining gene is the Z-bornepdgfa(with a chicken orthologue on GGA14). For the monitor lizard, the most promising candidate upstream sex-determining gene wasamh(anti Mullerian hormone), which (or the duplicated copy of which) is located on GGA28 and plays a conserved role in testis development in multiple teleost species (Herpin &Schartl, 2015), birds (Cutting et al., 2013), turtles (Zhou et al.,2019), and even the platypus (Zhou et al., 2021) and suggested to be a candidate sex determining gene in Komodo dragon (Lind et al., 2019). Intriguingly, the ortholog ofwt1(Wilms Tumour 1), an important regulator ofamhand master male-determining geneSryin human (Hossain & Saunders,2001), was determined to be X and Y-borne in the river turtle,and Z and W-borne in the monitor lizard, and was located on a secondary chicken site of GGA5 (Figure 5B; Supplementary Figure S2).
The expression patterns of these candidate sex-determining genes within the three reptiles for which we collected the gonad transcriptomes in this study further supported their function in the sex-determination pathway of each species(Figure 5C; Supplementary Figure S5). The Z-bornepdgfawas specifically expressed in the testis of the marbled gecko;whereas its downstream receptorpdgfra, which is X-borne in the river turtle, was strongly expressed in the ovary. The Xbornewt1of the river turtle, as well as the W-linkedwt1of the monitor lizard,were both expressed specifically in the ovary. It has to be noted that a previous study reported location ofwt1on to chromosome 1 in river turtle (Lee et al., 2019a), however ourin silicoanalysis identifiedwt1to be on X and Y chromosomes, which require further investigation. The Zbornewt1andamhof the monitor lizard were both specifically expressed in the testis. In summary, turnover of sex determining genes between the studied reptile species probably accounts for their sex chromosome turnovers.
Evolution of dosage compensation and sex-linked gene expression in the four reptile species
Having identified the sex chromosome-borne genes of the four distantly related reptiles, we set out to examine their diversity of dosage compensation based on comparison of gene expression levels between sexes, and between the autosomes and the sex chromosomes. Since sex chromosomes may undergo meiotic sex inactivation in germ cells, and gonads are probably not appropriate for direct comparison between sexes (Gu & Walters, 2017), we focused on comparing the expression levels between sexes in their somatic (brain, tail or blood) tissues. Genes that are shared between sex chromosomes are not expected to evolve dosage compensation. Also as mentioned earlier, we were unable to discriminate between the X- and Y-, Z- and W-borne genes (or called gametologs) based on their few divergence sites assembled from transcriptome sequences, we therefore focused on comparing the X- or Z-borne, or the hemizygous genes vs. autosomal genes for their somatic transcription level to examine the respective species’ dosage compensation pattern.
Among the four species, the worm lizard (with an XY sex system) and the monitor lizard (with a ZW sex system) have highly or moderately differentiated sex chromosomes. These two species exhibited a significantly (P<0.05, Wilcoxon test)different female vs. male expression ratio between autosomes and sex chromosomes (Figure 6A; Supplementary Figure S6).The X-borne genes were more female-biased in the worm lizard, and Z-borne genes were more male-biased in the monitor lizard, indicating incomplete dosage compensation in the two species. Similar patterns of incomplete dosage compensation have been reported before in Komodo dragon and pygopodidae lizards (Rovatsos et al., 2021). In contrast,genes on the undifferentiated sex chromosomes, as well as autosomes, of marbled gecko and river turtle showed no significant difference of their expression ratios between sexes.This could be because their Y- or W-borne genes have not degraded yet, so most genes on their X or Z chromosomes still have active partners on the Y (W) and thus there is no dosage difference between the sexes that selects for dosage compensation. In summary, all the four species examined do not seem to have evolved chromosome-wide dosage compensation mechanisms. On the other hand, for the monitor lizard with a much better assembly of its gametologs sequences, we further inspected their evolutionary rates(measured by ratios of nonsynonymous vs. synonymous substitution rates,dN/dS) using its related species Komodo dragon’s genome as a reference. Among the 52 gametologs present on both Z and W chromosomes, as expected, W gametologs exhibited a significantly (P<0.05, Wilcoxon test)faster rate of evolution than their Z-linked counterpart due to suppression of recombination (Figure 6B), indicating early signs of degeneration.
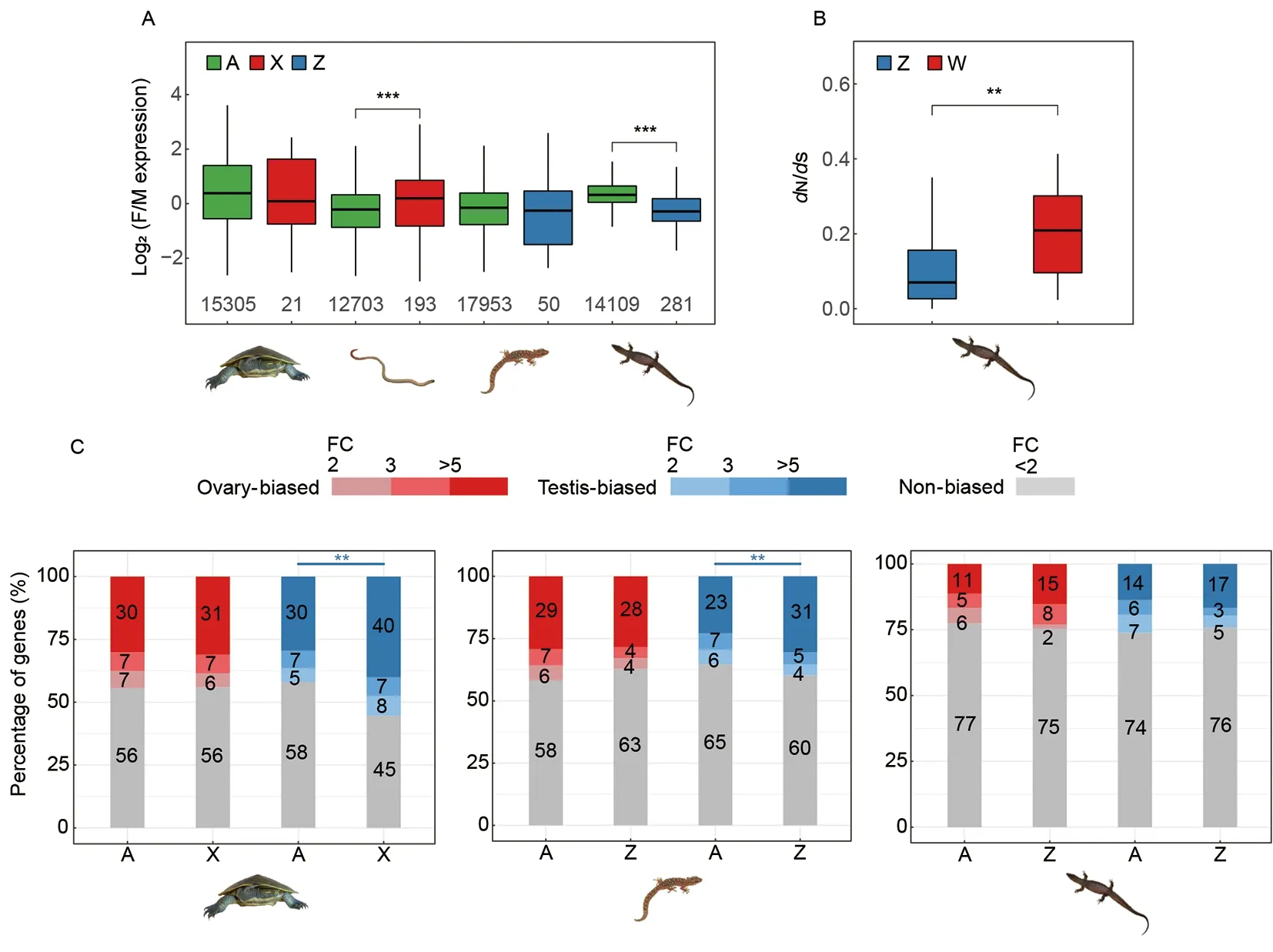
Figure 6 Dosage compensation and sex-linked gene expression in the four reptile species
For the three species with gonad transcriptomes (river turtle,marbled gecko and monitor lizard), we compared gene expression levels between sex chromosome vs. autosomes in the gonad, with the expectation that gonad-specific genes may have been preferentially selected to be located or not located on the sex chromosomes due to sex chromosomes’ sexbiased selective regimes. Previous studies inDrosophila(Assis et al., 2012) and other dipteran species (Vicoso &Bachtrog, 2015) found underrepresentation of male-biased or testis-biased genes, and overrepresentation of female-biased or ovary-biased genes on the X chromosome, supporting such sex-biased selective regime. We found that testis-biased genes were overrepresented (P<0.001,Chi-square test), while ovary-biased genes were underrepresented (P<0.005,Chisquare test) on the Z chromosome of the marbled gecko(Figure 6C). However, a similar masculinization and defeminization pattern was not found on the undifferentiated Z chromosome of the monitor lizard, probably because few Zborne genes were hemizygous (Figure 4).
The river turtle with undifferentiated XY sex chromosomes,unexpectedly showed a significant enrichment of testis-biased genes on the X chromosome relative to autosomal genes.This was probably because of cross-mapping of the reads of Y-borne genes that were not highly differentiated from those of X-borne genes. When we examined only the hemizygous Xlinked genes (those without a Y-linked homolog) there was no such enrichment pattern. This suggests some Y-borne genes of the river turtle have undergone a masculinization process even though they were still retained by the Y chromosome.
DISCUSSION
Given the large genomes of many reptile species (up to 5.3 Gb), fully sequencing sex chromosomes remains costly,despite the development of long-read sequencing and Hi-C technologies. So far, in depth studies of the gene content and dosage compensation of sex chromosomes have been carried out in a handful of lizards, snake species and turtle species(Alam et al., 2018; Bista et al., 2021; Ezaz et al., 2013;Rovatsos et al., 2021; Rupp et al., 2017; Schield et al., 2019;Vicoso et al., 2013) although ZW chromosome have been sequenced and a candidate sex determining gene identified in the central bearded dragon (Deakin et al., 2016).
Here, we developed a cost-effective method to expand our knowledge of sex-linked genes and sex chromosomes in a range of non-model reptiles and applied it to four distantly related reptile species. We used it to map sex chromosomeborne genes from male and female transcriptomes that were identified by screening with DNA probes from microdissected sex chromosomes. We also applied the novel stLFR linkedread sequencing technology (Wang et al., 2019) and assembled the draft genome of monitor lizard,V. acanthurus,including the sex chromosome sequences. The newly identified gene content of the sex chromosomes of these four distantly related reptile species provided new insights into reptile sex chromosome evolution and dosage compensation.
Previous studies showed that chicken and other birds have retained a very conserved karyotype close to that of reptilian ancestor (Deakin & Ezaz, 2019; Liu et al., 2021; Waters et al.,2021a). Mapping the chicken orthologues of sex chromosomeborne genes of the monitor lizard (V. acanthurus),worm lizard(A. parapulchella) and marbled gecko (C. marmoratus) onto the chicken genome revealed examples of recruitment of different ancestral autosomes. We found that the sex chromosomes of the monitor lizard (V. acanthurus),worm lizard (A. parapulchella) and marbled gecko (C. marmoratus)have homologues on different chicken autosomes. This implies that they evolved from different autosomes in a common reptilian ancestor.
However, our finding that sex chromosomes of the distantly related pink-tailed worm lizard and river turtle (E. macquarii)both have homology to GGA4q provides a striking example of convergent recruitment of ancestral autosome regions. The long arm of the chicken chromosome 4 (GGA4q) has also been previously reported to be recruited as sex chromosomes of pygopodid gecko (Rovatsos et al., 2021). This homology may signify that the same gene (likely to bepdgfra) has independently acquired a role in sex determination in all these species. Convergent recruitment of ancestral chromosome is a region orthologous to GGA23, which we identified to be part of the sex chromosomes of marbled gecko, and the central bearded dragon (Pogona vitticeps) (Ezaz et al., 2013) and among multiple species of turtles (Montiel et al., 2017).
Several general patterns emerged from these comparative analyses of the location of the chicken orthologues of genes on reptile sex chromosomes. Firstly, sex chromosomes seemed to have frequently originated by fusion of ancestral micro- and macro-chromosomes, or between microchromosomes (Waters et al., 2021b). In addition to homology to the chicken microchromosome GGA28 that was reported,and also confirmed in this work as the ancestral sex chromosome of Anguimorpha species including spiny tailed monitor lizard (Lind et al., 2019; Rovatsos et al., 2019), we found that other chicken microchromosomes GGA31, 33 contained fragments homologous to genes on the sex chromosomes of spiny tailed monitor lizard.Microchromosomes also seemed to have contributed to the sex chromosomes of three other reptiles (Figures 3, 4), as well as in the previously reported green anole lizard (Alföldi et al.,2011), bearded dragon lizard (Deakin et al., 2016), soft-shell turtles (Badenhorst et al., 2013; Kawagoshi et al., 2009). The short arm of chicken chromosome 4 (which is homologous to the conserved region of the X chromosome of therian mammals, is a microchromosome in all species other than the Galliformes. These observations of homologies with chicken microchromosomes are not surprising given that half the chicken genes lie on microchromosomes.
A microchromosome origin might have contributed to the second feature of reptile sex chromosomes, most of which are less differentiated than those of birds and mammals.Homomorphic or partially differentiated sex chromosomes were found in three out of four reptiles we examined and are also described also in the giant musk turtle (Kawagoshi et al.,2014), eyelid geckos (Kawagoshi et al., 2014; Pokorná et al.,2010) and some other gecko species (Koubová et al., 2014),and skinks (Kostmann et al., 2021). The preponderance of poorly differentiated sex chromosomes in reptiles could be the result either of slow differentiation, or rapid turnover, or both. A potential cause for the generally slower rate of sex chromosome differentiation in reptiles could be the high recombination rate and gene density of the ancestral microchromosomes (International Chicken Genome Sequencing Consortium, 2004), which might prevent extensive recombination suppression and rapid differentiation between sex chromosomes in these reptiles.
Alternatively, rapid turnover of reptile sex chromosomes could explain the “ever young” partially differentiated sex chromosomes that are so common in reptiles. We have previously demonstrated (Ezaz et al., 2009b) rapid transitions between sex determination systems in agamid lizards, and our present results expand the variety and independent origins of reptile sex chromosomes. In addition, the ability to switch into an environmental sex determination mode, and then to evolve novel genetic sex determination systems, may greatly facilitate turnovers. GSD and TSD have been reported within and between closely related reptile species, e.g., in agamid lizards(Ezaz et al., 2009a), in viviparous skink (Hill et al., 2018),some turtles (Bista & Valenzuela, 2020) and eye-lid geckos(Pensabene et al., 2020). In the Australian bearded dragon,the transition from GSD to TSD was observed both in the lab and in the field (Holleley et al., 2015), despite its possession of a pair of highly differentiated sex microchromosomes (Ezaz et al., 2005).
Our identification of genes on reptile sex chromosomes enabled us to assess their transcription and assess dosage compensation. We found no evidence of global dosage compensation, even in the worm lizardA. parapulchellawith highly differentiated X and Y chromosomes. This is similar to the absence of global dosage compensation in birds (Itoh et al., 2007) and reptiles (Rovatsos et al., 2021), but contrasts with the recently reported case of green anole lizard (Marin et al., 2017; Rupp et al., 2017), in which the single copy of the X chromosome is upregulated in XY males through an epigenetic mechanism similar to that inDrosophila. The absence of global dosage compensation inA. parapulchellacould reflect dosage mitigation or tolerance at posttranscriptional levels, or it may be a consequence of its dosage-dependent sex-determination mechanism, similar to that in chicken, in contrast to a male-dominant XY system of the green anole.
In this work we combined cytogenetics and high-throughput sequencing to characterize the sex chromosomes of four reptile species. This greatly widened our knowledge of sex chromosome birth, death and dosage compensation in a vertebrate class that shows particular variety in modes and turnover of sex determining systems.
Thus, we used DNA from microdissected sex chromosomes to identify transcripts of genes located on the XY or ZW chromosome pairs in each species, and located their chicken orthologues on different chicken chromosomes. This revealed the diverse origins of sex chromosomes, but detected convergent evolution between distantly related reptiles (turtle and worm lizard). Our novel pipeline efficiently identified candidate sex determining genes, which differed from those of birds and mammals. We found that none of the four species showed transcription profiles expected of global chromosomal dosage compensation.
In summary, our molecular and cytogenetic characterisation of sex chromosomes in diverse taxa greatly expands our knowledge of reptile sex determination. By identifying reptile candidate sex genes and providing the means with which to identify more, we hope to realise the value of this particularly variable, but understudied, vertebrate taxon, the only one for which no master sex determining gene has yet been discovered.
The inexpensive and efficient method developed here can be applied to studying any species of eukaryote with cytologically distinct sex chromosomes, providing the basis with which to better understand the ecological and evolutionary drivers of sex chromosomes and sex determination systems.
DATA AVAILABILITY
The genomic and transcriptomic data of worm lizard (A.parapulchella),river turtle (E. macquarii), marbled gecko (C.marmoratus) and monitor lizard (V. acanthurus) have been deposited in NCBI under BioProjectID PRJNA737594. The draft genome and annotation of monitor lizard (V. acanthurus)have been deposited in Genome Warehouse (GWH) under BioProject accession No. PRJCA005583. All the data have been deposited to Science Data Bank with data CSTR:31253.11.sciencedb.j00139.00016 and 31253.11.sciencedb.01919.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
T.E., K.M., J.A.M.G. conceived the idea. K.M., F.S., J.D.conducted lab works. Z.X.Z. and Q.Z. conducted bioinformatic analyses. Q.Z., Z.X.Z. and T.E. wrote the first draft. All coauthors contributed intellectually to writing and editing the draft multiple times. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
Authors would like to acknowledge feedback by Janine Deakin on a preliminary draft. Animal photo credit: worm lizard and marbled gecko-T.G.; monitor lizard-J.D.; river turtle-A.G. and chicken-Liesl Taylor.

- Zoological Research的其它文章
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
- The Hippo pathway and its correlation with acute kidney injury
- Genomics and morphometrics reveal the adaptive evolution of pikas
- Molecular phylogeny and taxonomy of four Remanella species (Protozoa, Ciliophora): A flagship genus of karyorelictean ciliates, with descriptions of two new species