
Phylogenetic analysis of combined mitochondrial genome and 32 nuclear genes provides key insights into molecular systematics and historical biogeography of Asian warty newts of the genus Paramesotriton(Caudata: Salamandridae)
2022-10-17 03:27TaoLuoShaShaYanNingXiaoJiaJunZhouXingLiangWangWeiCaiChenHuaiQingDengBaoWeiZhangJiangZhou
Zoological Research 2022年5期
Tao Luo, Sha-Sha Yan, Ning Xiao, Jia-Jun Zhou, Xing-Liang Wang, Wei-Cai Chen, Huai-Qing Deng,Bao-Wei Zhang, Jiang Zhou,*
1 School of Life Sciences, Guizhou Normal University, Guiyang, Guizhou 550025, China
2 School of Karst Science, Guizhou Normal University, Guiyang, Guizhou 550001, China
3 Guiyang Healthcare Vocational University, Guiyang, Guizhou 550081, China
4 Zhejiang Forest Resource Monitoring Center, Hangzhou, Zhejiang 310020, China
5 Key Laboratory of Environment Change and Resources Use in Beibu Gulf Ministry of Education, Nanning Normal University, Nanning,Guangxi 530001, China
6 School of Life Sciences, Anhui University, Hefei, Anhui 230601, China
ABSTRACT The Paramesotriton Chang, 1935 genus of Asian warty newts is the second most diverse genus in the family Salamandridae, currently containing 14 recognized species from northern Vietnam to southwest-central and southern China. Although species of this genus have been included in previous phylogenetic studies, the origin and interspecific relationships of the genus are still not fully resolved,especially at key nodes in the phylogeny. In this study, we sequenced mitochondrial genomes and 32 nuclear genes from 27 samples belonging to 14 species to reconstruct the interspecific phylogenetic relationships within Paramesotriton and explore its historical biogeography in southern China. Both Bayesian inference and maximum-likelihood analyses highly supported the monophyly of Paramesotriton and its two recognized species groups (P. caudopunctatus and P. chinensis groups)and further identified five hypothetical phylogenetic cryptic species. Biogeographic analyses indicated that Paramesotriton originated in southwestern China (Yunnan-Guizhou Plateau/South China)during the late Oligocene. The time of origin of Paramesotriton corresponded to the second uplift of the Himalayan/Qinghai-Xizang (Tibetan) Plateau(QTP), rapid lateral extrusion of Indochina, and formation of karst landscapes in southwestern China.Principal component analysis (PCA), independent sample t-tests, and niche differentiation using bioclimatic variables based on locations of occurrence suggested that Paramesotriton habitat conditions in the three current regions (West, South,and East) differ significantly, with different levels of climatic niche differentiation. Species distribution model (SDM) predictions indicated that the most suitable distribution areas for the P. caudopunctatus and P. chinensis species groups are western and southern/eastern areas of southern China. This study increases our knowledge of the taxonomy,biodiversity, origin, and suitable distribution areas of the genus Paramesotriton based on phylogenetic,biogeographic, and species distribution models.
Keywords: Paramesotriton; Systematics;Biogeography; Southern China
lNTRODUCTlON
Mountain ecosystems often show high species richness and diversity due to a combination of geology and climate(Antonelli et al., 2018; Hazzi et al., 2018; Rahbek et al.,2019a, 2019b). Southern China has long been considered as a little-known and highly threatened biodiversity hotspot and a priority area for biodiversity conservation by the Chinese government (Hu et al., 2021; Ministry of Environmental Protection, 2015; Myers et al., 2000). High diversity and regional endemism provide the opportunity to trace the roles of complex geological and climatic histories in species diversity. In recent years, a number of biogeographic studies have explored the evolutionary history of different ecotypes in southern China, including cave fish (Wen et al., 2022),amphibians and reptiles (Che et al., 2010; Chen et al., 2018a;Guo et al., 2020; Hofmann et al., 2019; Wang et al., 2018a; Xu et al., 2021; Yan et al., 2021), invertebrates (Zhao & Li, 2017),and plants (Xiang et al., 2021; Zhao et al., 2021). However,little is known about the origin and dispersal of salamanders,such as Asian warty newts, in this region.
TheParamesotritonChang, 1935 genus of Asian warty newts is the second most diverse genus within the family Salamandridae, containing 14 recognized species distributed in mountain streams and rivers in southern China and northern Vietnam (AmphibiaChina, 2022; Frost, 2021).Species inParamesotritonare highly dependent on riverine habitats and typically have a narrow distribution due to their low dispersal ability and reliance on water bodies (Fei et al.,2006; Zhao et al., 2012). Thus, species within this genus are ideal for exploring the evolutionary history and drivers of speciation of aquatic fauna. However, populations continue to decline due to habitat loss as a result of environmental pollution, climate change, and anthropogenic disturbance(e.g., dam construction, tourism, pet trade, and agriculture)(IUCN, 2022; Zhao et al., 2012). For example,P.guangxiensis,P. zhijinensis, andP. yunwuensisare considered endangered (IUCN, 2022), with wild populations currently threatened with extinction and requiring conservation attention. Short-term evolutionary histories of species (e.g.,recent species formation and adaptive radiation) exhibiting high morphological similarity can mislead assessment of species diversity and evolutionary relationships, leading to poor conservation decisions. Therefore, molecular analyses are required to elucidate the species diversity, evolutionary relationships, and conservation units inParamesotriton(Luo et al., 2021).
Unstable and uncertain phylogenetic relationships may affect our understanding of diversity and evolutionary history.The phylogenetic position ofParamesotritonwithin the family Salamandridae has been resolved (Kieren et al., 2018; Zhang et al., 2008), but relationships withinParamesotritonremain poorly understood, especially between species groups(Dubois & Raffaëlli, 2009; Dubois et al., 2021; Fei et al., 2006;Fei & Ye, 2016; Gu et al., 2012a; Yuan et al., 2014;Supplementary Figure S1 and Table S1). Fei et al. (2006)divided ChineseParamesotritonspecies into theP.caudopunctatusandP. chinensisspecies groups based on morphological characters, which were accepted by later researchers (Gu et al., 2012a, 2012b; Wang et al., 2013; Wu et al., 2010; Yuan et al., 2014, 2016a; Zhao et al., 2008).Subsequently, Fei & Ye (2016) divided ChineseParamesotritonspecies into the three subgeneraAllomesotriton,Karstotriton, andParamesotritonbased on skeletal and morphological characters (Supplementary Table S1). Several recent phylogenetic studies have attempted but failed to resolve the relationship between the two species groups (Gu et al., 2012a; Yuan et al., 2014, 2016a).Furthermore, current taxonomic hypotheses regarding the relationships within and between the two species groups (or three subgenera) depend on morphological characters and single fragments of mitochondrial DNA (mtDNA) sequences.Therefore, expanded sample collection and additional molecular markers are required for phylogenetic analysis to resolve the interspecific relationships and evolutionary history ofParamesotriton.
Herein, based on the most complete and expanded sampling to date, the phylogeny and biogeography of the genusParamesotritonwas inferred using mitochondrial and nuclear locus data, with results used to analyze environmental variables based on occurrence data. The study objectives were to (1) explore the interspecific relationships of the genusParamesotriton, and (2) identify the center of origin, estimate dates of each divergence event, and reconstruct the evolutionary history of the genus.
MATERlALS AND METHODS
Taxon sampling and sequence collection
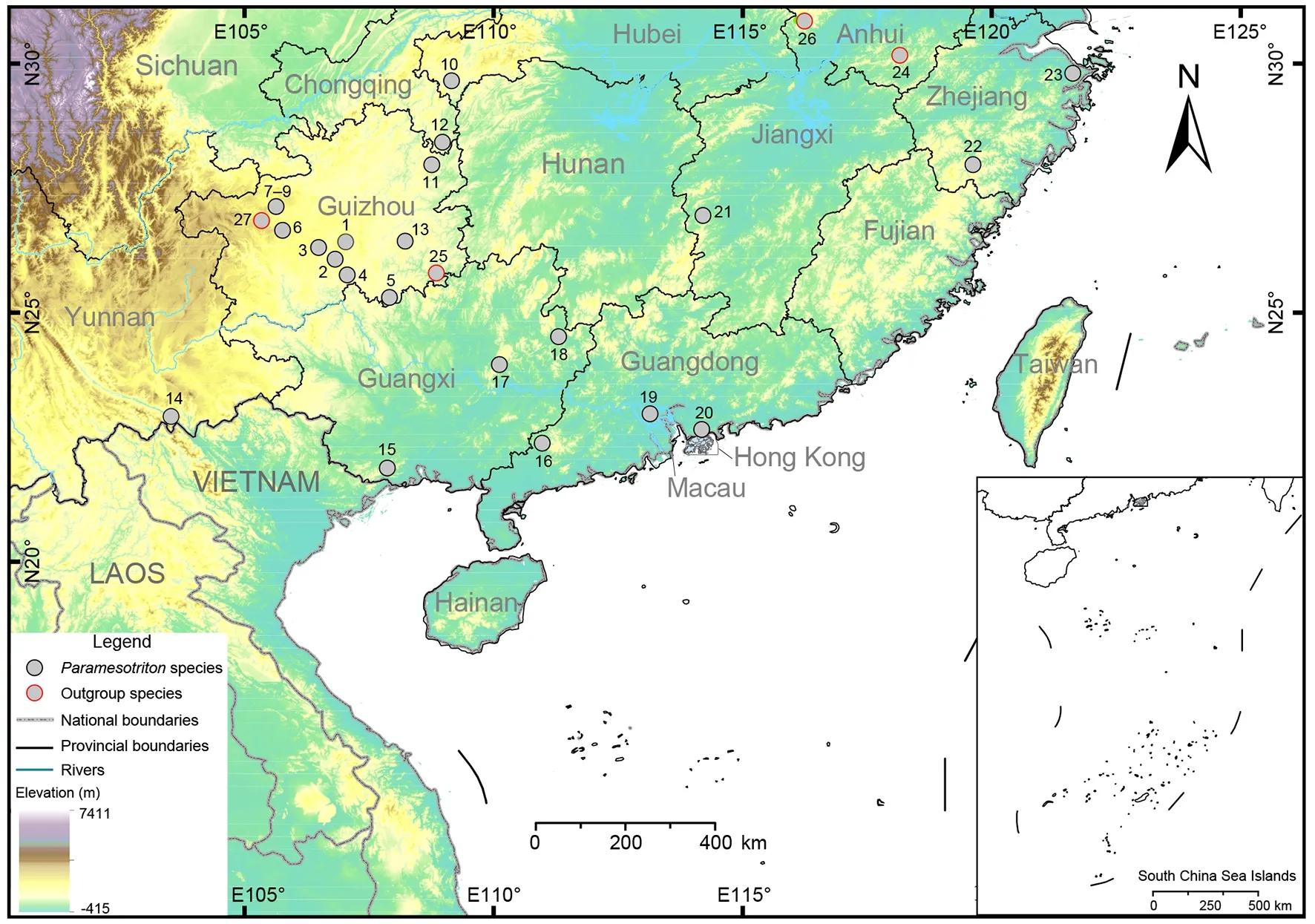
Figure 1 Locations of 14 species of Paramesotriton and four outgroup samples collected for this study
A total of 23Paramesotritonsamples were collected from Guizhou, Zhejiang, Guangxi, Guangdong, Hubei, Yunnan,Jiangxi, and Hong Kong, China (Figure 1), representing 14 recognized species and putative cryptic species. We also searched the National Center for Biotechnology Information(NCBI, https://www.ncbi.nlm.nih.gov/) database using“Paramesotritonmitochondrion, complete genome” as keywords (as of 7 January 2022) and downloaded the mitochondrial genome (mitogenome) of the genus for phylogenetic analysis (see Supplementary Table S2 for more details). Following Zhang et al. (2008), we selected
Pachytriton feii,Pachytriton inexpectatus,Cynops orientalis,Tylototriton kweichowensis, andTylototriton asperrimusas outgroups for phylogenetic analysis. Species names,localities, specimen vouchers, and accession numbers of outgroups and ingroups are provided in Supplementary Table S2. All samples for molecular analysis are kept at the Guizhou Normal University, Guiyang City, Guizhou Province, China.
DNA extraction, amplification, and sequencing
Genomic DNA for each sample was extracted from 95%ethanol-preserved tissue using the cetyltrimethylammonium bromide (CTAB) method (Allen et al., 2006). For each sample,DNA was cleaved into small fragments (350 bp) using ultrasound. The end DNA fragments were repaired using 3'-5'nucleic acid exonuclease and polymerase; a single base “A”was added at the 3' end, and sequencing connectors were ligated. The DNA fragments were then selected by agarose gel electrophoresis and polymerase chain reaction (PCR)amplification was performed to form a sequencing library.Sequencing was performed on an Illumina NovaSeq 6000 sequencer. At the end of sequencing, high-quality clean reads were obtained using SOAPnuke v1.3 (Chen et al., 2018b) by the removal of reads with more than 5% N base content, lowquality (≤5) bases up to 50%, and adapters. Ultimately, each sample generated reads that were 150 bp in length and ~4.2 Gb in size. The reads were assembled into mitogenomes using SPAdes v3.14.0 (Bankevich et al., 2012). Assembled contigs were BLAST aligned with the relevant reference genome (P. caudopunctatus; EU880326) to identify potential assembly errors. Finally, the included genes in the assembled mitogenomes were annotated using MITOS v2.0 (Bernt et al.,2013).
To obtain a stable phylogenetic framework and resolve interspecific relationships, we also amplified 32 nuclear gene fragments for inclusion in the phylogenetic analysis. All samples were amplified using PCR under the following conditions: initial denaturation step at 98 °C for 3 min; 36 cycles of denaturation at 95 °C for 30 s, annealing at 51-59°C for 40 s, extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. PCR primers and detailed annealing temperatures for the 32 nuclear gene fragments are provided in Supplementary Table S3. The purified PCR products were directly sequenced in both directions using an ABI Prism 3730 automated DNA sequencer from TSINGKE Biotechnology Co.,Ltd. (Chengdu, China). All newly obtained mitogenomic and nuclear gene sequences were deposited in GenBank under accession Nos. MW524141-MW524143, ON357921-ON357943, ON364656-ON365439, and ON422325(Supplementary Table S2).
Phylogenetic analysis
The newly obtained mitogenome and nuclear gene sequences were edited using PhyloSuite v1.2.2 (Zhang et al., 2020) and DNAStar (DNASTAR, Inc., USA). Sequence alignment was performed using MAFFT v7.4 (Katoh & Standley, 2013) with default settings in PhyloSuite v1.2.2. The resulting sequences were checked, and ambiguous alignments were trimmed by eye using MEGA v7.0 (Kumar et al., 2016). For the mitogenome, we selected 39 gene fragments for phylogenetic analysis, i.e., two ribosomal genes (12S, 16S), 13 mitochondrial protein-coding genes (ATP6,ATP8,COI,COII,COIII,ND1,ND2,ND3,ND4,ND4L,ND5,ND6, and cytb),and 24 transfer RNAs (tRNAs). The sequence matrix used for phylogenetic analysis consisted of concatenated mitochondrial data and 32 nuclear loci data. The best-fit partitioning scheme and corresponding nucleotide substitution models for concatenated nucleotides were selected using PartitionFinder v2.1.1 (Lanfear et al., 2017) based on Bayesian information criterion (BIC).
Phylogenetic analysis of concatenated nucleotide sequences was performed under the best-fit partitioning schemes and nucleotide substitution models using Bayesian inference (BI) and maximum-likelihood (ML) in MrBayes v3.2.1(Ronquist et al., 2012) and IQ-tree v2.0.4 (Nguyen et al.,2015), respectively. ML analysis was run using the best-fit model for each partition with 2 000 ultrafast bootstrap (UFB)replicates (Minh et al., 2013) and was performed until a correlation coefficient of at least 0.99 was reached (Hoang et al., 2018). BI analysis was run independently using four Markov Chain Monte Carlo (MCMC) chains (three heated chains and one cold chain) starting with a random tree; each chain was run for 2×107generations and sampled every 1 000 generations. Convergence of data runs was estimated using average standard deviation of split frequencies (ASDSF)<0.01 and Tracer v1.7.1 (Rambaut et al., 2018) to check for effective sample size (ESS) >200. The phylogenetic tree nodes were considered well-supported when the Bayesian posterior probability (BPP) of the node was ≥0.95 and ML UFB was ≥95%. Using MEGA v7.0 (Kumar et al., 2016), the uncorrectedP-distance model (1 000 replicates) was used to calculate the average genetic distance between mitogenomes of species.
Divergence-time estimation
We used BEAST v2.6.6 (Bouckaert et al., 2014) to estimate the date of origin of the genusParamesotritonand time of divergence of each species using concatenated nucleotide data under a relaxed clock log-normal assumption and ana prioriYule model. In this analysis, three calibration point constraints were used: (1) divergence between primitive newts(Tylototriton) and modern Asian newts (Cynops,Paramesotriton, andPachytriton) dated to 74.2 million years ago (Ma) (Zhang et al., 2008), with a normal prior (mean 74.2;sigma 5.0; offset 0.0); (2) origin of modern Asian newts calibrated to 15 Ma based on nearly complete early Miocene fossils ofProcynops miocenicus, which are clearly related toCynops, with a log-normal prior (mean 10; sigma 1.0; offset 15) and calibration set to monophyly (Estes, 1981; Kieren et al., 2018); and (3) divergence between theP. caudopunctatusgroup andP. chinensisgroup of the genusParamesotritondated to 23.8 Ma (95% HPD: 22.4-35.0 Ma; Zhang et al.,2008), with a normal prior (mean 23.8; sigma 2.0; offset 0.0).
BEAST analysis was conducted for a total of 2×107generations, with sampling every 2 000 generations. We used Tracer v1.7.1 (Rambaut et al., 2018) to check the convergence and stability of run parameters, i.e., ESS of parameters >200. Maximum clade credibility (MCC) trees were generated using TreeAnnotator v2.4.1 (Rambaut &Drummond, 2010) by applying a burn-in (as trees) of 10%,posterior probability limit of 0.5, and median height for node selection. Time tree editing and visualization were performed in FigTree v1.4.3 (Rambaut, 2016).
Ancestral area reconstruction
To estimate the origin and dispersal history ofParamesotriton,BioGeoBEARS (Matzke, 2013) in RASP v3.2 (Yu et al., 2015)was used to reconstruct ancestral areas and infer their biogeographic history. A time-calibrated phylogenetic tree generated from BEAST analysis was used as the input tree with all outgroups removed, retaining onlyParamesotritonspecies. Based on the natural landscape, zoogeography proposed by Zhang (2011), and current distribution ofParamesotriton, we used three distribution areas: i.e., (A)Yunnan-Guizhou Plateau; (B) South China/North Vietnam;and (C) East China. Considering the environmental dependence ofParamesotritonspecies, we assumed that extant species would not occur in more than two distribution areas. Therefore, the maximum number of species range areas was limited to two.
Ecological niche modeling, differentiation, and environmental variation analysis
In this study, published and web-recorded data were collected between 2000 and 2021 via multiple means (Supplementary Table S4). If distribution information was only available for location, OvitalMap v9.3.4 was used to infer the coordinates of the location centroid. Records with obvious geocoding errors or where relative accuracy could not be determined were discarded, and duplicate records were manually removed. We use ENMTools v1.3 (Warren et al., 2010) to filter occurrence data based on environmental variable layers to avoid overfitting of prediction results. We collected a total of 165 recorded sites for species in the genusParamesotriton,including 70 recorded sites in theP. caudopunctatusspecies group and 95 recorded sites in theP. chinensisspecies group(East 44, South 51).
Species distribution models (SDMs) for both species groups were predicted using the maximum entropy model implemented in Maxent v3.3.1 (Phillips et al., 2006). The 19 bioclimatic variables (Supplementary Table S5) (resolution 2.5 arc-min) were downloaded from the WorldClim database as environmental data (http://www.worldclim.org, Hijmans et al.,2005a). In SDM analysis, high multicollinearity among bioclimatic variables (Peterson & Nakazawa, 2008),regularization multipliers, and feature classes (Low et al.,2021) were the main factors affecting the prediction results(Radosavljevic & Anderson, 2014). We use two methods to reduce this impact. First, because high multicollinearity between bioclimatic variables may improve SDM accuracy, we used correlation analysis (Pearson’sr>0.85; Dormann et al.,2013) in ENMTools (Warren et al., 2010) to minimize correlation between variables. Second, different regularization multipliers (0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, and 2.0)and feature classes were assessed in R using the “kuenm”package (Cobos et al., 2019) to obtain the best combination of runs in Maxent (Phillips et al., 2006; Phillips & Dudík, 2008).Candidate model performance was evaluated based on significance (partial receiver operating curve (ROC), 500 iterations, and 50% of data for bootstraps), omission rate(E=5%), and model complexity (AICc). The best models were selected using on the following criteria: (1) significant model,(2) omission rate ≤5%, and (3) final model delta AICc ≤2 based on best set of models obtained from (1) and (2) (Cobos et al., 2019). As a result, low multicollinearity (Supplementary Figure S2) Bio1 (Annual Mean Temperature), Bio4(Temperature Seasonality), Bio6 (Min Temperature of Coldest Month), Bio13 (Precipitation of Wettest Month), Bio17(Precipitation of Driest Quarter), Bio19 (Precipitation of Coldest Quarter), regularization multiplier 1.2 (P.caudopunctatusspecies group)/0.2 (P. chinensisspecies group East or South), and feature class selected linearquadratic features (P. caudopunctatusandP. chinensisspecies groups South/East) were used for SDM analysis(Supplementary Table S6).
Based on the best parameters and models (Supplementary Table S7), to reduce uncertainty caused by sampling artifacts,we randomly divided the distribution data into training data(75%) and validation data (25%) in Maxent. To evaluate the robustness of the models, subsampling was repeated 10 times and the distribution points were repeatedly split into random training and testing subsets, after which the results were averaged. The maximum number of background points and iterations were set to 10 000 and 5 000, respectively, to find the optimal solution, with default values used for the remaining parameters (Hazzi et al., 2018; Morales et al., 2017; Phillips et al., 2006; Radosavljevic & Anderson, 2014). The generated suitability maps were in ASCII format and were visualized using ArcMap v10.4 (Esri). At the same time, we reclassified the suitability layers into four classes based on the predicted results: i.e., unsuitable habitat (0.00-0.25), minimally suitable habitat (0.25-0.50), moderately suitable habitat (0.50-0.75),and highly suitable habitat (>0.75). To explore the key climatic variables that shape the distribution of each species, jackknife tests were performed and response curves for each bioclimatic variable were plotted. To verify the accuracy of each model, we used area under the curve (AUC) (Allouche et al., 2006; Swets, 1988) of the ROC (Hanley & McNeil, 1982)based on following criteria: poor (AUC<0.8), good (AUC 0.90-0.95), and excellent (AUC>0.95) (Guisan & Thuiller,2005).
Ecological niche overlap of species within the twoParamesotritonspecies groups was measured using occurrence site and spatial environmental data (Broennimann et al., 2012). Climatic niche equivalency among and within species groups (forP. chinensisspecies group) inParamesotritonwas estimated based onI(Warren et al.,2008) andDstatistics (Schoener, 1968). Analysis was performed using “ecospat” in the R package with the“niche.equivalency.test” function (Di Cola et al., 2017).Observed values ofDandIwere compared to 100 pseudoreplicates, with both statistics ranging from 0 to 1(Warren et al., 2008). The observed values ofDandIwere significantly lower than the niche similarity distribution of 100 replicates and were significantly different (P<0.05), indicating niche differentiation (Warren et al., 2008). In addition, to test whether the ecological niches obtained for the twoParamesotritonspecies groups were identical, climatic niche similarity analysis was conducted using the“ecospat.niche.similarity.test” function in the “ecospat”package in R, i.e., comparing actual occurrence of one species group with random background occurrence of another species group. Here, for niche equivalency and similarity analyses, 100 replicate niche equivalency and background tests were conducted.
Point-based analysis was used to test for differences in bioclimatic variables of the habitats of species from the two species groups in the West, South, and East regions of southern China. Values of the 19 bioclimatic variables for 165 occurrence locations were extracted using DIVAGIS v7.5(Hijmans et al., 2005b). Principal component analysis (PCA)and independent samplet-tests (1 000 bootstrap replicates)were used to test whether differences in ecological niches between the West, South, and East regions were statistically significant.
RESULTS
Nucleotide sequence information
In this study, 27 mitogenomes and 784 sequences from 32 nuclear loci were obtained by sequencing (including outgroups) (Supplementary Table S2). We downloaded seven mitogenomes and 30 nuclear gene sequences from GenBank(Supplementary Table S2). The combined mtDNA (15 408 bp)included 5 774 variable sites and 4 623 parsimony-informative sites. Alignment of concatenated nuclear DNA (nuDNA)contained a total of 27 023 bp, including 1 827 variable sites and 1 150 parsimony-informative sites. Finally, we obtained a matrix of concatenated mtDNA and nuDNA sequences with a total length of 42 431 bp. Detailed information of each sequence dataset and best-fit nucleotide substitution model for each partition are listed in Supplementary Table S8.
Phylogenetic inference
The phylogenetic trees obtained from the concatenated mtDNA and nuDNA datasets using BI and ML analyses showed similar topologies (Figure 2). Both BI and ML analyses highly supported the monophyly ofParamesotritonand the monophyly of theP. caudopunctatus(Clade A) andP.chinensisspecies groups (Clade B) (BPP=1.00; UBP=100%).TheP. caudopunctatusspecies group consisted of five species, i.e.,P. caudopunctatus,P. wulingensis,P.zhijinensis,P. maolanensis, and four populations ofP.longliensis(Longli, Huishui, Dafang, and Hubei populations)and could be further divided into two subclades. Subclade A1 consisted ofP. caudopunctatusandP. wulingensis, and subclade A2 consisted ofP. zhijinensis,P. maolanensis, andP. longliensis. TheP. chinensisspecies group consisted of nine species, i.e.,P. fuzhongensis,P. yunwuensis,P.guangxiensis,P. deloustali,P. labiatus,P. qixilingensis,P.chinensis,P. aurantius, andP. hongkongensis, and could be further divided into three subclades. Subclade B1 consisted of

Figure 2 Phylogenetic tree reconstructed using Bl and ML methods based on concatenated mtDNA and nuDNA datasets for the genus Paramesotriton and outgroups
P. fuzhongensis,P. yunwuensis,P. guangxiensis, and two populations ofP. deloustali. Subclade B2 consisted ofP.labiatusand subclade B3 consisted ofP. qixilingensis,P.chinensis,P. aurantius, and two populations ofP.hongkongensis.
The average mtDNA-based uncorrectedP-distances between and within species ofParamesotritonare provided in Table 1, with interspecific genetic distances ranging from 0.52% (P. maolanensisvs.P. longliensis(LL)) to 11.84% (P.zhijinensisvs.P. labiatus). Within subclade A1 of theP.caudopunctatusspecies group, averageP-distances ranged from 0.52% to 1.83%, with averageP-distances of 2.19% and 1.05% within theP. hongkongensisandP. deloustalipopulations, respectively (Table 1).
Spatiotemporal analysis based on concatenated mtDNA and nuDNA data
The time tree generated from the BEAST run was consistent with the phylogenetic trees inferred by BI and ML, except for subclade A. Spatiotemporal maps of divergence time and ancestral area reconstructions are shown in Figure 3. The date of the stem node ofParamesotritonwas estimated at 25.42 Ma (95% highest posterior density (HPD): 21.27-29.79 Ma). The time to the most recent common ancestor of the genusParamesotritonand the split between theP.caudopunctatusandP. chinensisspecies groups was estimated at 22.86 Ma (node 1; 95% HPD: 19.38-26.45 Ma)(Figure 3B). Early lineage diversification ofParamesotritonoccurred in the mid-Miocene (15.69-10.19 Ma; nodes 2-6).Lineage diversification of theP. caudopunctatusandP.chinensisspecies groups occurred in the mid- to late Miocene(15.69-5.94 Ma) and Pliocene/Pleistocene boundary(3.06-1.28 Ma).
Comparisons of the six models within BioGeoBEARS using corrected Akaike information criterion (AICc) weighting are listed in Table 2. Results showed that the fit of the two remaining “ +J” models, but not the DEC+J model, was significantly better compared to the models without “+J ”,indicating the importance of founder event speciation in explaining biogeographic patterns (Table 2). According to the best-fit model DIVALIKE+J (Figure 3B), both vicariance and dispersal events may have acted as the main driving forcesshaping the current geographic distribution patterns of the genusParamesotriton.
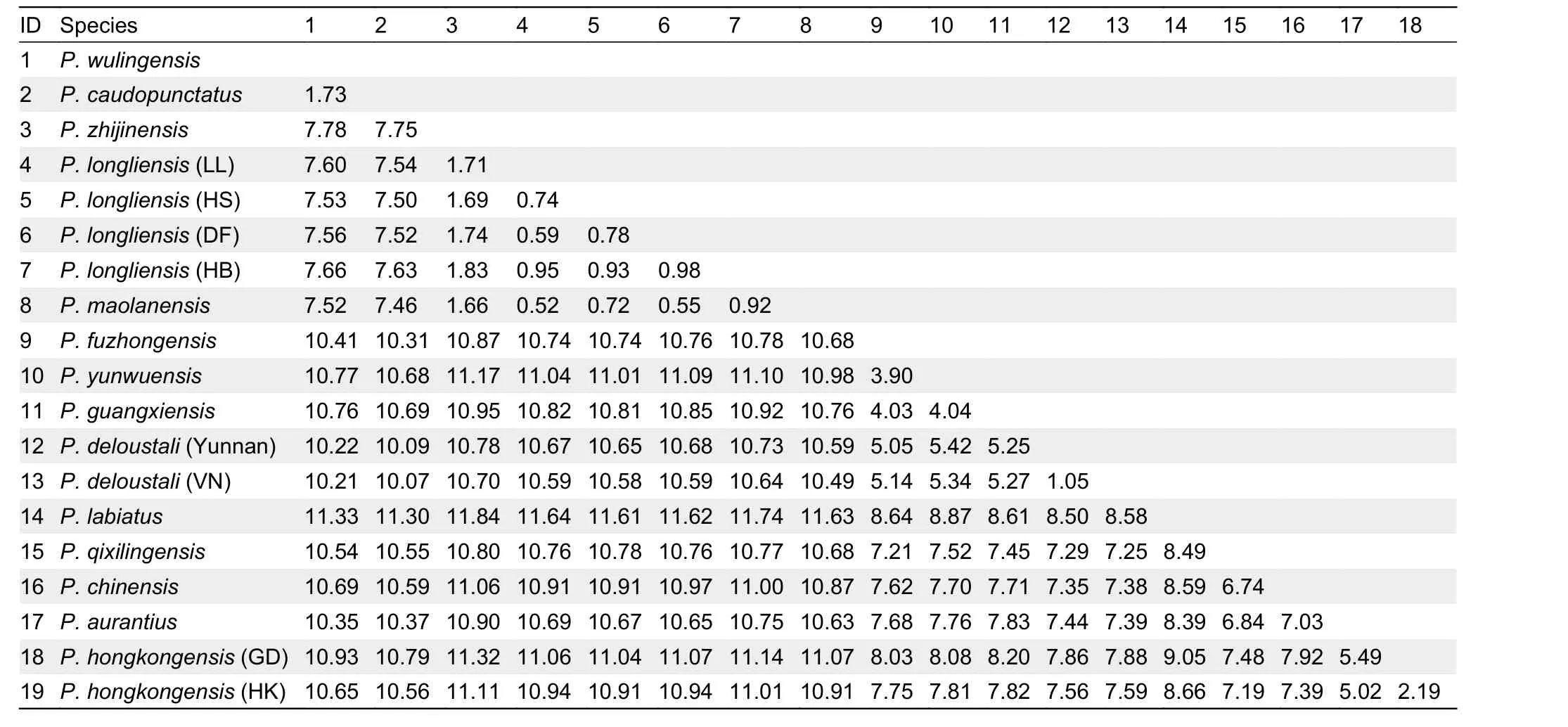
Table 1 Uncorrected mean P-distances (%) between Paramesotriton species on mitogenome
The BioGeoBEARS results for the best model(DIVALIKE+J) suggested that the recent ancestor of extant species of the genusParamesotritonwas most likely distributed on the Yunnan-Guizhou Plateau/South China and dispersed into Guizhou in South China after a vicariance event(Figure 3B; node 1). TheP. caudopunctatusspecies group originated from the Yunnan-Guizhou Plateau (mainly Guizhou), and theP. chinensisspecies group originated from southern China. At least four dispersal events and two vicariance events occurred in the mid-Miocene (12.81-8.65 Ma; nodes 6-7). During the mid-Miocene (12.81 Ma; node 6),dispersal events from southern to eastern China initiated the formation and expansion of subclade B3 (Figure 2). During the Miocene (8.65 Ma; node 7), a dispersal event from eastern to southern China (Hong Kong and Guangdong, China) was the origin of the formation ofP. hongkongensis. Finally, the current geographic distribution patterns within theP.caudopunctatusandP. chinensisspecies groups were formed by 13 and 12 speciation events, respectively.
Suitable distribution, climatic niche differentiation, and environmental variation analysis
The SDMs generated good predictions of occurrence locations under current climate scenarios. The AUCs for theP.caudopunctatusspecies group (West),P. chinensisspecies group (East), andP. chinensisspecies group (South) across the observation region were 0.988, 0.987, and 0.991,respectively. The most suitable habitats for theP.caudopunctatusspecies group (West) were in the karst mountain ecosystems of Guizhou in southwestern China, and moderately suitable habitats were located in the Wuling Mountains (southeastern Chongqing, northwestern Hunan,and western Hubei) (Figure 4A). In the East, suitable habitats for theP. chinensisspecies group (East) were found in Fujian,Zhejiang, Jiangxi, and Anhui in eastern China and in the Nanling Mountains (southern Hunan, and southern Jiangxi)and Luoxiao Mountains (Figure 4B). In the South, suitable habitats for theP. chinensisspecies group (South) were found in Guangdong, Hong Kong, southern Guangxi, and southern Fujian (Figure 4C).
Species within the twoParamesotritonspecies groups exhibited different levels of niche differentiation (Figure 5).Climatic niche equivalency analysis indicated that bothIandDvalues (<0.1) were close to 0 (Figure 5A-F), suggesting that the niches between theP. caudopunctatus(West) andP.chinensisspecies groups (East),P. caudopunctatus(West)andP. chinensisspecies groups (South), andP. chinensis(East) andP. chinensisspecies groups (South) were close to non-overlapping (Figure 5M-P). Niche similarity tests showed that the null hypothesis of identical niche occupation was rejected between theP. chinensis(East),P. chinensis(South),andP. caudopunctatusspecies groups (West), suggesting that the pairs occupied different niches, although this was not strongly supported (P>0.05, Figure 5G, H, K, L). The spatial similarity of occupied niches between theP. caudopunctatus(West) andP. chinensisspecies groups (East) was more similar than the simulated random distribution but was not significantly supported (P>0.05, Figure 5I, J).
PCA of the 19 bioclimatic variables generated four principal components (PC1=56.39%, PC2=24.41%, PC3=11.18%, and PC4=5.57%), which explained 96.55% of total variance in ecological niche differences under the current climate scenario(Table 3). The PC1 and PC2 scatter plots are shown in Figure 4D, which clearly separated theP. caudopunctatus(West),P. chinensis(East), andP. chinensisspecies groups(South). Independent samplet-tests suggested that of the 19 bioclimatic variables, 10 were significantly different between West and East, 12 were significantly different between West and South, and eight were significantly different between East and South (P<0.05, Table 4).
The stepmother, however, took this letter as well, and wrote a new one, in which the king ordered that the queen and the two little princes should be burnt at the stake
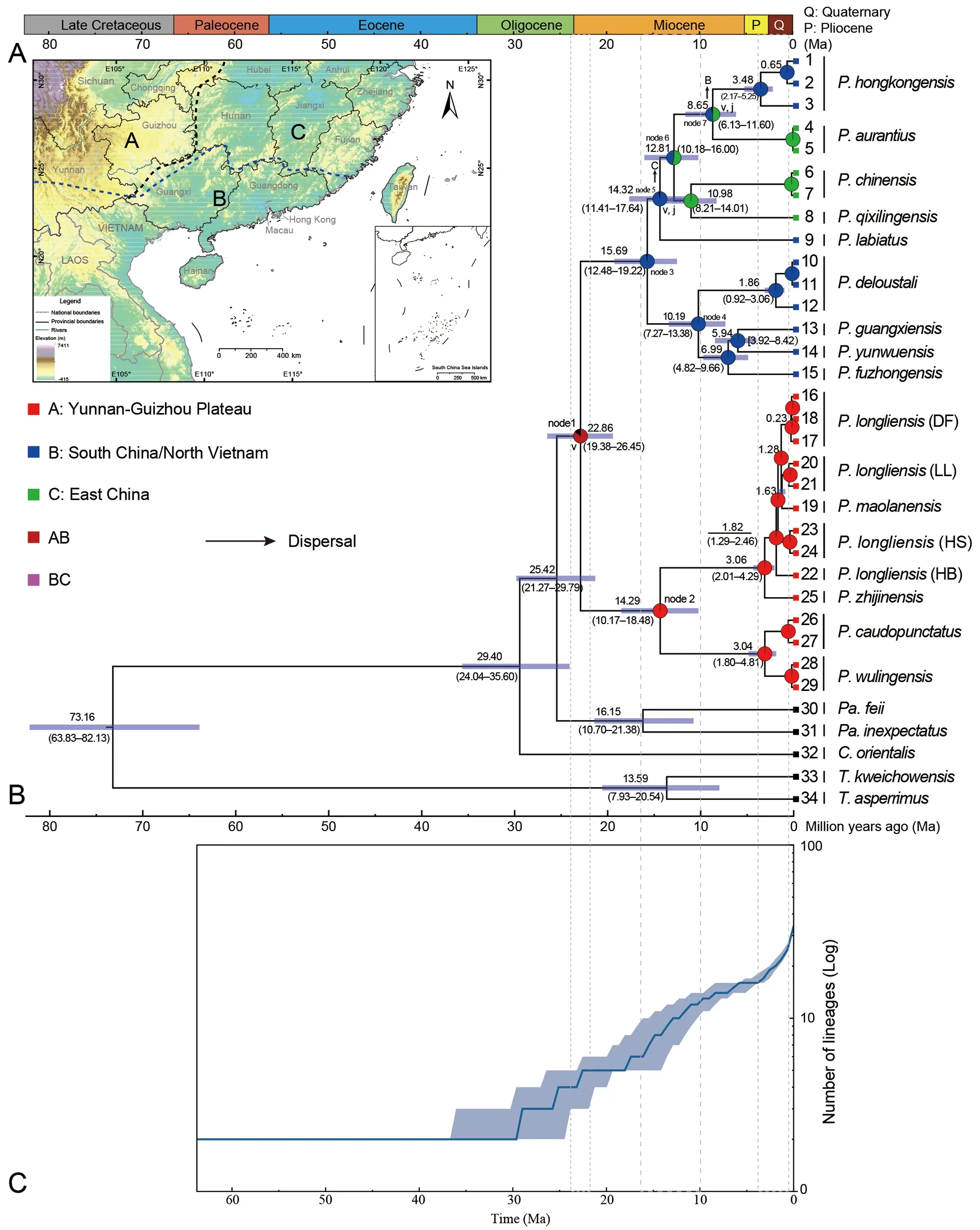
Figure 3 Distribution areas (A) and time-calibrated topology of the genus Paramesotriton, reconstructed ancestral area of Paramesotriton based on best-fit model (DlVALlKE+J) using BioGeoBEARS (B), and lineage-through-time (LTT, logarithmic scale) plot (C)
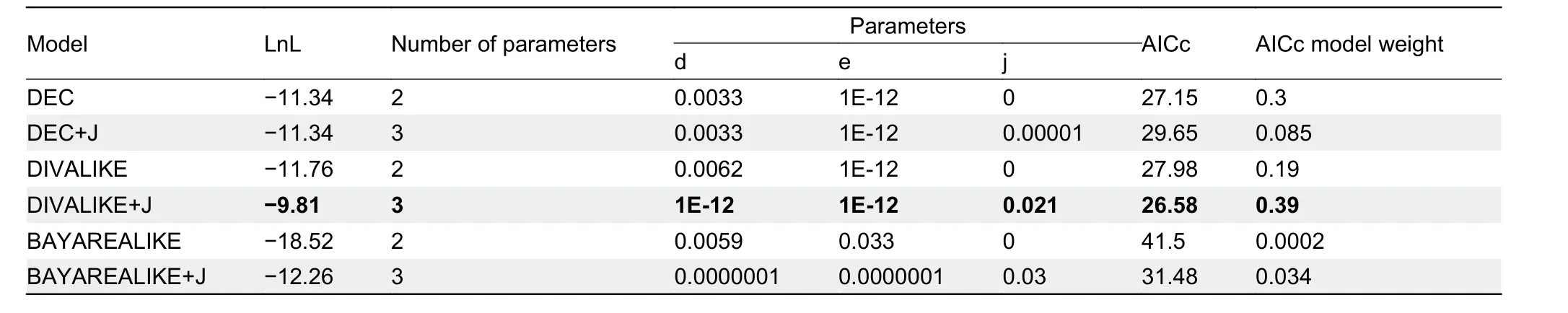
Table 2 BioGeoBEARS estimations of ancestral areas based on phylogeny of the genus Paramesotriton
DlSCUSSlON
Phylogeny and classification
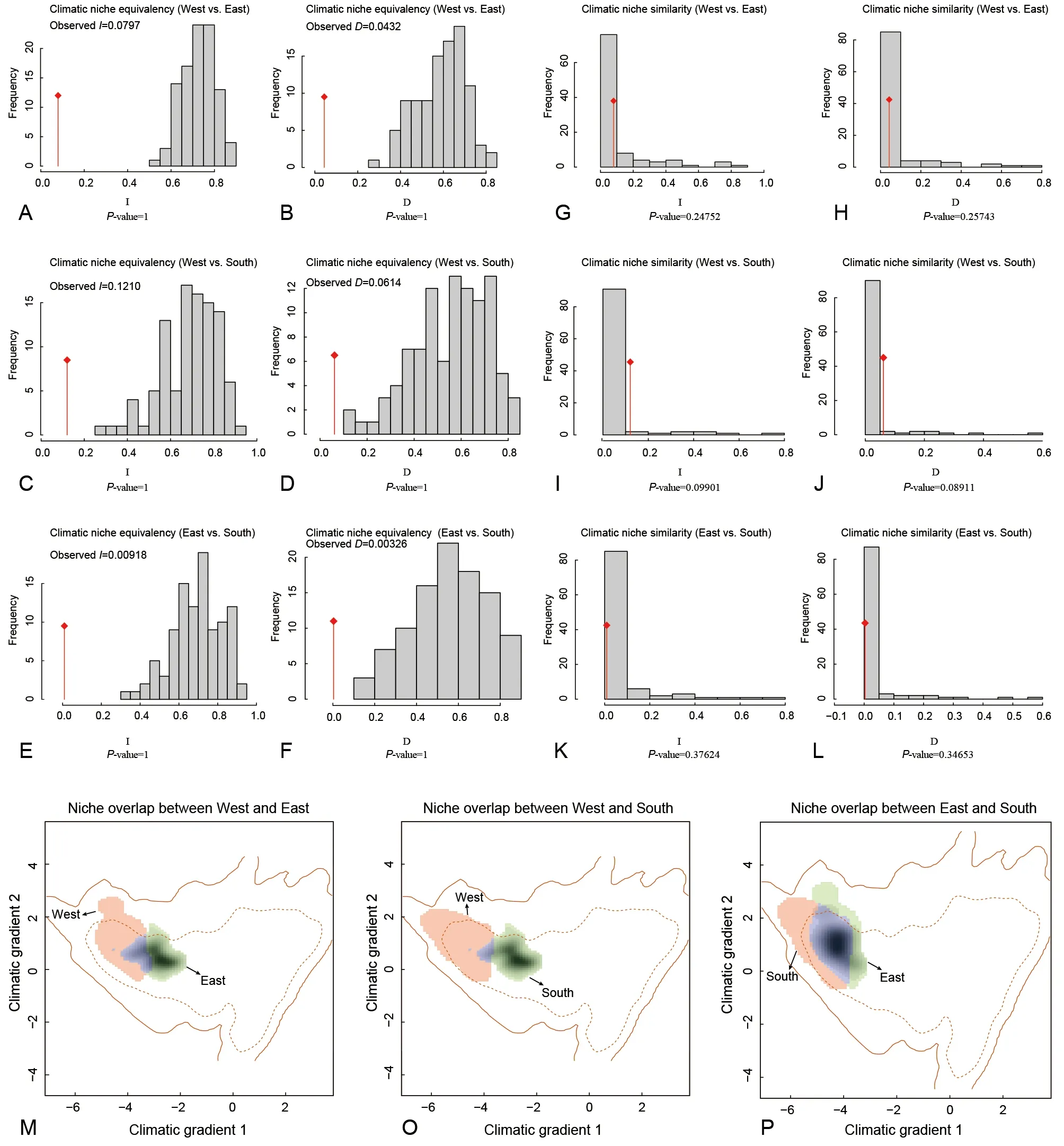
Figure 5 Climatic niche differentiation of P. caudopunctatus species group (West) and P. chinensis species group (East/South) in Paramesotriton in southern China
Here, mitogenomes and 32 nuclear loci were concatenated into a data matrix for phylogenetic analysis of the genusParamesotriton. The amount of molecular data and number of samples analyzed exceeded all previous phylogenetic studies of the genus (Chan et al., 2001; Dubois et al., 2021; Gu et al.,2012a; Lu et al., 2004; Yuan et al., 2014; Zhang et al., 2008).Our analysis resolved a long-standing phylogenetic controversy concerning species groups and interspecific relationships (Figure 2). Overall, phylogenetic analysis highly supported theP. chinensisspecies group (subgenus
Paramesotriton) andP. caudopunctatusspecies group(subgeneraAllomesotritonandKarstotriton) previously proposed as monophyletic based on morphological characters
(Fei et al., 2006; Fei & Ye, 2016).
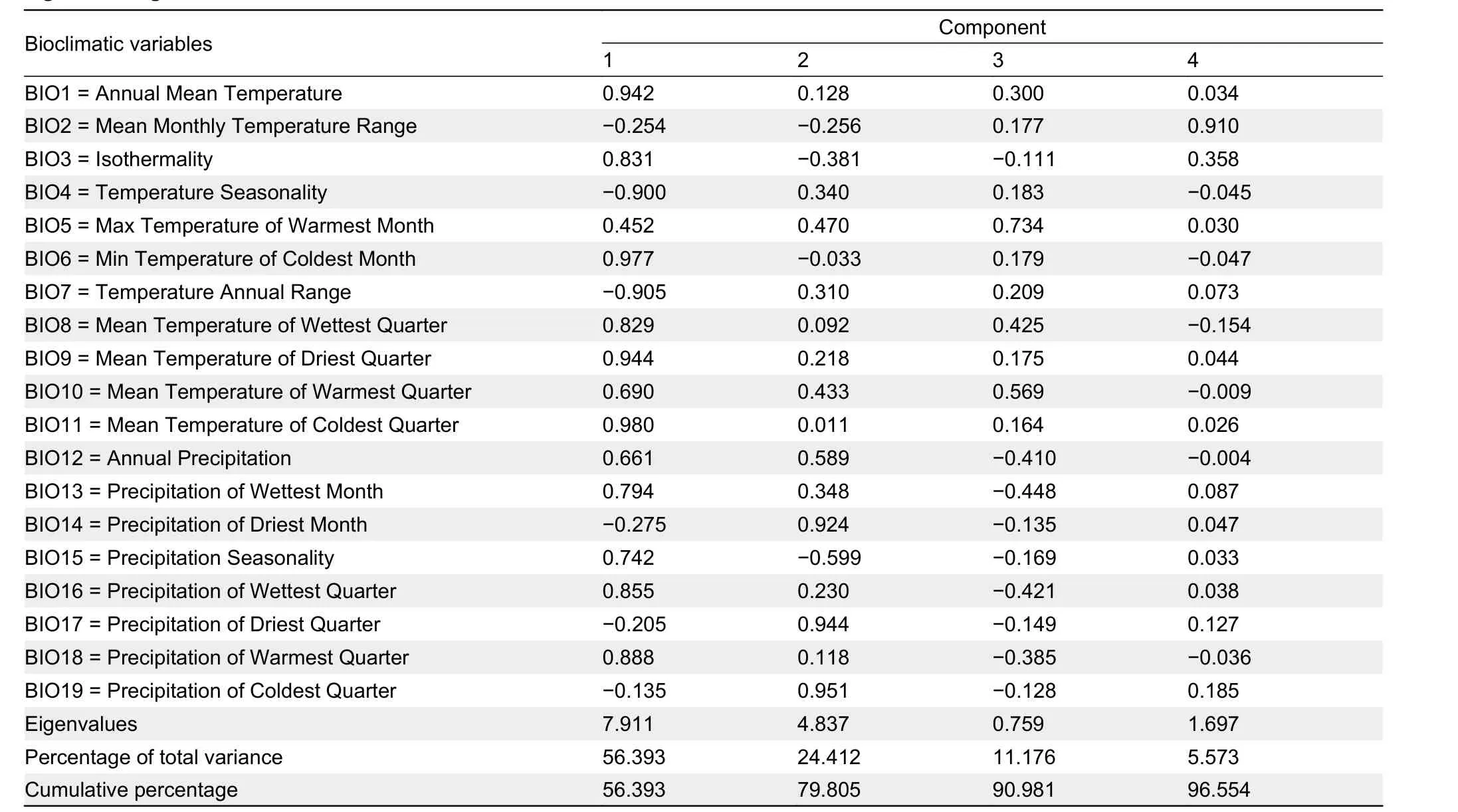
Table 3 Nineteen bioclimatic variables were used for principal component analysis (PCA) based on occurrence locations and eigenvalues greater than one

Table 4 lndependent sample t-tests based on 19 bioclimatic variables for occurrence locations of P. caudopunctatus and P. chinensis species groups
Within theP. caudopunctatusspecies group, the phylogenetic relationships of species were well resolved, and the obtained topology was similar to previous studies (Gu et al., 2012a, 2012b; Luo et al., 2021; Yuan et al., 2014, 2016a).Due to the position ofP. labiatus, the obtainedP. chinensisspecies group phylogeny was inconsistent with previous tree topologies (Yuan et al., 2014, 2016a). Inferred from mitochondrial partial tRNAMet,ND2, and flanking tRNAs(tRNATrpand partial tRNAAla), the most likely position ofP.labiatuswas as sister taxon to all remaining members (P.chinensisandP. hongkongensis) of theP. chinensisspecies group except forP. guangxiensis,P. deloustali, andP.fuzhongensis(Wu et al., 2009; Supplementary Figure S1F),although this result was only weakly supported by partitioned ML, maximum parsimony, and BI. Using mitochondrialND2and flanking tRNAs (tRNATrpand partial tRNAAla), Yuan et al.(2014) identifiedP. labiatusas the sister taxon to all members of theP. chinensisspecies group with very high support.Using a similar molecular marker and partitioning strategy, Wu et al. (2010) and Yuan et al. (2016a) found that the phylogeny of theP. chinensisspecies group contained three polytomic clades, withP. labiatusat the bottom (Supplementary Figure S1I). Notably, although these studies shared the same molecular markers and similar analytical methods, except for number of sequences, the evolutionary models were different.In this study, the phylogenetic relationships within theP.chinensisspecies group were subclade B1 ((P. deloustali+(P.guangxiensis+(P. yunwuensis+P. fuzhongensis)))+(subclade B2 (P. labiatus)+subclade B3 ((P. qixilingensis+P.chinensis)+(P. aurantius+P. hongkongensis))), whileP.labiatuswas identified as a sister taxon to the remaining species of theP. chinensisspecies group identified in Yuan et al. (2014) or was unresolved (Yuan et al., 2016a). The reason for this was due to differences in the type and number of molecular markers (e.g., Yuan et al., 2014, 2016a only used mitochondrialND2and its flanking tRNAs), as reflected in other amphibian studies (Wu et al., 2020; Yan et al., 2021). In conclusion, compared to previous studies (Wu et al., 2009,2010; Yuan et al., 2014, 2016a), our phylogenetic tree provides good resolution of the interspecific phylogenetic positions of species within the genusParamesotriton.
Hidden species diversity
Cryptic species may still be present inParamesotriton. For example, Li et al. (2008a, 2008b) and Gu et al., (2012b)
describedP. longliensis,P. zhijinensis, andP. maolanensis
Historical biogeography
In East Asia, breakthroughs in amphibian and reptile biogeography in the last decade suggest that geological and climatic changes since the Cenozoic have influenced biodiversity patterns in southern China (Che et al., 2010; Chen et al., 2018a, 2020; Guo et al., 2019, 2020; Hofmann et al.,2019; Li et al., 2013; Liang et al., 2018; Oliver et al., 2015;Yuan et al., 2016b, 2018; Wu et al., 2020; Zhou et al., 2021).However, comparatively little attention has been paid to tailed amphibians, especially salamanders. Geological changes in East Asia may have influenced the early evolutionary history ofParamesotriton. Dating analyses indicated thatParamesotritonoriginated about 25.42 Ma (95% HPD:21.27-29.79 Ma) near the late Oligocene (Cohen et al., 2013),followed by early diversification and vicariance into two species groups during the early Miocene (ca. 22.86 Ma). This period coincides with geological events at the Oligocene/Miocene boundary, i.e., rapid lateral extrusion of Indochina (Tapponnier et al., 1990; Wang et al., 2006; Zhao et al., 2016), uplift of the Himalayan/QTP (An et al., 2006; Bai et al., 2010; Ding et al., 2017; Mulch & Chamberlain, 2006; Sun,1997), and formation of karst landscapes in southwestern China (Che & Yu, 1985). The timing of this origin and estimated date of interspecific divergence are consistent with co-distributed species in East Asia, such as amphibians (Che et al., 2010; Chen et al., 2013; Wu et al., 2020; Zeng et al.,2020), reptiles (Guo et al., 2020), plants (Deng et al., 2018;Sun et al., 2022; Xiang et al., 2016, 2021; Zhao et al., 2016),and invertebrates (Zhang & Li, 2013; Zhao & Li, 2017).
Subsequent lineage diversification ofParamesotritonduring the mid-Miocene closely correlated with geological movement and climate change during that period. Diversification of the main lineages of the twoParamesotritonspecies groups first peaked in the mid-Miocene (nodes 2-6; Figure 3C), a period characterized by further rapid uplift of the Himalayan/QTP (ca.20-10 Ma; Ding et al., 2017; Favre et al., 2015) and global climate decline (Miao et al., 2012; Westerhold et al., 2020;Zachos et al., 2001). The uninterrupted uplift of the eastern edge of the QTP (e.g., Hengduan Mountains) and the Wuyi-Nanling Mountains from the mid-Miocene (ca. 15-10 Ma)significantly altered the mountain landscape and river erosion patterns in the region (Favre et al., 2015; López-Pujol et al.,2011; Yan et al., 2018). This orogeny drove the establishment of the mountain and modern river systems, e.g., the Pearl River, Minjiang River, and lower Yangtze River (Yan et al.,2018; Zheng et al., 2013; Zheng, 2015), possibly contributing to lineage diversification ofParamesotriton, especially theP.chinensisspecies group. Orogeny in small regions may also have caused species diversification. Within theP.caudopunctatusspecies group, the mid-Miocene (node 2,Figure 3B) was divided into subclades A1 and A2 (Figure 2).The main geological event during this period was the equalization of the action of the Philippine Sea and Indian plates on the South China/Southeast Asian plate, which led to the slow uplift of the crust to form a low mountainous landscape with high western and low eastern edges,accompanied by regional-scale orogenic movements,including the formation of the ancient Wuling Mountains (Zhou& Chen, 1993). Geographical isolation due to the Wuling Mountains may have led to divergence of theP.caudopunctatusspecies group into two subclades, while the subsequent formation and westward development of the Miaoling in the early Pleistocene (Lin, 1993; Zhou & Chen,1993) together with the warm and humid climate (Bureau of Geology and Mineral Guizhou Province, 1987; Ji, 1992; Li,2001; Tong et al., 1994) likely promoted species diversification of the group. Thus, the combined effects of geology and climate change in East Asia from the mid-Miocene likely promoted diversification of species withinParamesotriton.Similar patterns of diversification have been found in other Asian amphibians, such as the Asian leaf-litter frog genusLeptobrachella(Chen et al., 2018a), Asian torrent frog genusAmolops(Wu et al., 2020; Zeng et al., 2020), and Asian knobby newt genusTylototriton(Wang et al., 2018a). This common pattern suggests that amphibians distributed in southern China may have exhibited common evolutionary responses to the geological and climatic changes that occurred during the Miocene. Finally, the formation ofParamesotritonand the Yangtze River coincided closely, with species diversifying subsequent to the formation of the river(Favre et al., 2015; Fu et al., 2020; Zheng et al., 2013; Zheng,2015). Thus, the low dispersal ability of the genus, geographic isolation of the Yangtze River, and limited movement ability of the species may be the main factors limiting the spread ofParamesotritonspecies north of the Yangtze River.
The rich species diversity (Hu et al., 2021; Myers et al.,2000), high endemism (Lei et al., 2015; Ma et al., 2019; Wang et al., 2017), and natural environment of southwestern China make it an important area for biologists studying the origins of species diversity in East Asia. Ancestral area reconstruction and spatiotemporal analysis suggested that the ancestors of the genusParamesotritonmay have originated in the karst areas of southwestern China (Yunnan-Guizhou Plateau/South China), as reported previously for cave fish (Wen et al., 2022;Zhao & Zhang, 2009), frogs (Ye & Fei, 2001; Yuan et al.,2016b; Zhou et al., 2017), and snakes (Guo et al., 2020).According to the current geographic distribution and phylogenetic tree, we propose a “vicariance-dispersalvicariance” hypothesis for the evolutionary history ofParamesotriton. The highly heterogeneous habitats created by the intense uplift of the QTP, rapid lateral extrusion of Indochina, and karst landscape formation led to the isolation and splitting of ancestral species ofParamesotritoninto the West and South/East clades. The West Clade, corresponding to theP. caudopunctatusspecies group, contributed to species formation in and around Guizhou after dispersal and vicariance. The ancestors of the East Clade, corresponding to theP. chinensisspecies group, dispersed from southern to eastern China and contributed to species formation after vicariance. The subsequent East-to-South dispersal event also explains the narrow sympatric distribution of several species in the east. In addition, considering the special geographic location and long natural evolutionary history of southwestern China, the West-South-East geographic distribution and phylogenetic patterns suggest a West-to-East dispersal pattern forParamesotriton(Figures 1, 3). As this dispersal pattern has also been found in some snakes (Guo et al., 2011, 2016, 2020) and frogs (Yan et al., 2021; Yuan et al.,2016; Zhou et al., 2017) in southern China, a West-to-East dispersal pattern may be widely present in other taxa in southern China, although there are some exceptions showing East-to-West dispersal (Liang et al., 2018).
We note that recent evolutionary radiation may have occurred in theP. caudopunctatusspecies group, triggering rapid adaptation to the karst environment, and accelerating the formation of cryptic species. However, genomic studies are needed to reveal the complete evolutionary history and adaptive mechanisms.
Environmental heterogeneity and spatial distribution
Environmental factors play important roles in driving speciation, spatial distribution, and adaptation (Antonelli et al.,2018; Pereira & Wake, 2009; Vieites et al., 2007). To understand the phylogenetic differences in geographic distribution from West-to-East and North-to-South, we tested the influence of environmental heterogeneity on the spatial distribution of salamander species in southern China. Based on bioclimatic variables, PCA, independent samplet-tests,and niche differentiation analysis revealed significantly different habitat environmental conditions and some climatic ecological niche differentiation among the three regions(Table 3; Figure 4). In terms of physical geography, theParamesotritondistribution area belongs to the eastern monsoon region of China (Zhang, 2011), while the western and eastern regions belong to the mid-subtropics (including the coasts of Zhejiang and Fujian reaching westward to the Yunnan-Guizhou Plateau) and the southern part belongs to the south subtropics (including southern Fujian, Guangxi,Guangdong, and Yunnan), which differ significantly in terms of climate, rainfall, and topography. Climatically, southwestern and southeastern China are influenced by the warm and wet monsoon from the Indian Ocean and the Asian monsoon from the Pacific Ocean (An et al., 2001; Jiang & Ding, 2009; Ren et al., 2021; Vornlocher et al., 2021; Wan et al., 2007).Zoogeographically, in southern China, there are different divisions between the eastern (Eastern Hillock Plain subregion) and western (Western Mountainous Plateau subregion) parts of Central China of the Oriental Realm, and the southern part of South China (Min’guang Coastal sub-region)(Xing et al., 2008; Zhang, 2011). Geomorphologically, the West region is a fully developed, high-coverage karst landscape, whereas the East region is almost karst-free, and the South region is a poorly covered karst landscape(Goldscheider et al., 2020; Zhang, 1980). Ecological differentiation and environmental adaptation are considered to play important roles in driving species differentiation and formation (McCulloch et al., 2021; Nosil, 2012; Rundle & Nosil,2005; Wang et al., 2018b). Here, the species distribution model predictions suggested that theP. caudopunctatusandP. chinensisspecies groups only have suitable habitat in West, East, and South China (Figure 4). The combination of these factors may reflect adaptive responses of salamanders to the microhabitat of the location of occurrence in the current climatic scenario. Although we still know little about the adaptive mechanisms ofParamesotritonin different ecoregions in southern China at the morphological and genetic levels, it appears that the western and eastern landscapes and resulting ecological changes are closely correlated with the evolution of West-to-East divergence (Guo et al., 2020; Yan et al., 2021). Future research should focus on the adaptation ofParamesotritonto karst water environments, conservation biology, and population genetic diversity, which are important for understanding the adaptation of amphibians to water environments and global climate change.
CONCLUSlONS
We investigated the phylogeny, biogeography, environmental variation, and geographic distribution patterns of the mountain salamander genusParamesotritonbased on a sample set covering 14 species, combined with mitogenomic and nuclear gene data. The phylogeny and monophyly of the genus and its two species groups,P. caudopunctatusandP. chinensisspecies groups, were determined, and five phylogenetic cryptic species were identified. Biogeographic analyses suggested thatParamesotritonoriginated in southwestern China (Yunnan-Guizhou Plateau/South China) in the late Oligocene, with subsequent dispersal from South China northward to East China, forming West-South-East geographic distribution patterns in terrains II and III in South China. Based on bioclimatic variation and interspecific geographic distribution patterns, we suggest that the natural landscape of karst mountains in southern China and changes in ecological factors played important roles in the evolution ofParamesotritonfrom West-to-East and the formation of the West-South-East distribution pattern.
SClENTlFlC FlELD SURVEY PERMlSSlON lNFORMATlON
All samples were obtained following Chinese regulations for the Implementation of the Protection of Terrestrial Wild Animals (State Council Decree (1992) No. 13), and the Guidelines for the Care and Use of Laboratory Animals by the Ethics Committee at Guizhou Normal University (Guiyang,Guizhou, China).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
J.Z. and T.L. conceived and designed the research; T.L.,Y.S.S., N.X., J.J.Z., X.L.W., W.C.C., and B.W.Z. conducted field surveys and collected samples; T.L., N.X., and B.W.Z.performed molecular work; T.L. H.Q.D., and S.S.Y. analyzed molecular and environmental data; T.L., Y.S.S., J.Z., and H.Q.D. wrote, discussed, and revised the manuscript. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We are grateful to Prof. Zhi-Yong Yuan, Prof. Xiao-Ming Gu,and Dr. Xiao-Long Liu for providing tissue samples used for molecular analysis. We thank Kai Gao, Xiang Zeng, Jun Zhou,Hong-Tao Cui, Xiang-Di Fan, Ya-Li Wang, and Si-Wei Wang for their help during sample collection. We thank LetPub(www.letpub.com) for its linguistic assistance during manuscript preparation.

- Zoological Research的其它文章
- Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
- The Hippo pathway and its correlation with acute kidney injury
- Genomics and morphometrics reveal the adaptive evolution of pikas