
Metagenomic analysis reveals presence of different animal viruses in commercial fetal bovine serum and trypsin
2022-10-17 03:27PengZhangLeCaoYingYingMaBinSuChiYuZhangYanPengLi
Zoological Research 2022年5期
Peng Zhang, Le Cao, Ying-Ying Ma, Bin Su, Chi-Yu Zhang,*, Yan-Peng Li,*
1 Shanghai Public Health Clinical Center, Fudan University, Shanghai 201508, China
2 Beijing Key Laboratory for HIV/AIDS Research, Beijing Youan Hospital, Capital Medical University, Beijing 100069, China
ABSTRACT Animal-derived biological products, such as fetal bovine serum (FBS) and trypsin, are important supplements for scientific, pharmaceutical, and medical use. Although preventive guidelines and tests are implemented to reduce potential viral contamination in these biologicals, they do not target unusual or emerging viruses, leading to safety concerns. Using unbiased metagenomics, we investigated the presence of viruses in recently collected commercial FBS and trypsin samples from different geographic regions. In total, we detected viral sequences belonging to Parvoviridae,Anelloviridae, Flaviviridae, Herpesviridae,Caliciviridae, Nodaviridae, Rhabdoviridae, and Paramyxoviridae, including several viruses related to bovine diseases, viruses of potential human and insect origin, and viruses of unknown origin. Bovine parvovirus 3 and bosavirus were detected with high frequency and abundance in FBS, necessitating more stringent testing for these parvoviruses during production. Both bovine norovirus and bovine viral diarrhea virus 1 displayed relatively high genetic distance to closest hits, indicating the presence of new genotypes in farm animals. While the origin of novel lyssavirus and Nipah virus is unclear, their presence raises the possibility of the introduction of pathogenic animal-derived viruses into biologicals.Our results showed relatively widespread contamination of different viruses in biologicals,underscoring the need for robust safety protocol alternatives, such as metagenomic sequencing, to monitor emerging viruses.
Keywords: Virome; FBS; Trypsin; Biosafety;Viral contaminations
INTRODUCTION
Samples of animal origin are widely used to produce biological products for scientific and medical applications. For example,bovine serum and trypsin (mostly bovine or porcine origin) are extensively applied in cell culture for vaccine production,in vitrodiagnostics, gene therapy, stem cell therapy, and food production (Paim et al., 2021). However, these biological products may carry animal or even human-related viral elements during the production process, and viral contamination may impact the safety of cell banks and production of subsequent biological products for medical use(Gagnieur et al., 2014). Increasing viral contamination of cell culture reagents for scientific research has also been identified(Porter et al., 2021). Whether the use of contaminated biological products produces misleading results is also of concern, e.g., the stimulation of specific cell signaling pathways and detection of animal viruses in human samples.
Animals are important vectors and/or reservoirs of emerging and re-emerging viruses, such as rabies virus, Nipah virus,Ebola virus, Zika virus, H1N1 influenza virus, West Nile Virus,dengue virus, cowpox virus, severe acute respiratory syndrome coronavirus, and Middle East respiratory syndrome coronavirus (Grange et al., 2021; Harvey & Holmes, 2022;Wille et al., 2021). Some viruses circulate in animals and only cause asymptomatic infections in their natural hosts. However,some viruses can lead to serious disease or can jump to new hosts and cause zoonotic events (Dong & Soong, 2021;Grange et al., 2021). The wide use and transportation of animal-origin samples may introduce pathogenic viruses into new populations of previously unexposed animals, leading to the spread of veterinary epidemics (Marcus-Sekura et al.,2011).
Routine standards and tests have been established for biological production to control contamination and reduce potential transmission and safety risks, e.g., 9 CFR e Code of Federal Regulations from the US Department of Agriculture(USDA) and EMA/CHMP/BWP/457920/2012 rev.1/2 Regulations from the European Medicines Agency (Gagnieur et al., 2014; Paim et al., 2021). Bovine and porcine-related viruses are commonly detected in bovine serum and trypsin(Baylis et al., 2020; Chen et al., 2008; Pinheiro De Oliveira et al., 2013; Sadeghi et al., 2017; Wang et al., 2018), which are the most widely used biological products worldwide.However, viral removal measures, such as inactivation and filtration, are not required for all commercial sample batches(Paim et al., 2021), and certain virus types, such as parvoviruses, are less sensitive to these traditional measures(Sadeghi et al., 2017). Animals carry far more viruses than reported. Even for known viruses, current regulatory lists for biologicals (serum and trypsin) cover only limited virus types,including bovine adenovirus, bovine parvovirus (BPV), bovine respiratory syncytial virus, bovine viral diarrhea virus (BVDV),rabies virus, reovirus, bluetongue virus, porcine adenovirus,porcine circovirus, porcine parvovirus transmissible gastroenteritis virus, and porcine hemagglutinating encephalomyelitis virus (Marcus-Sekura et al., 2011).However, the use of biological products may pose potential safety issues for viruses not included in current regulatory lists.
With the rapid growth in viral discovery using metagenomic sequencing, an increasing number of viral sequences are available in public databases. Continuous surveillance of viruses carried in biological products will help reduce contamination issues during research experiments, identify potential origins of viruses, and discover emerging and/or neglected viruses. In the present study, we used unbiased viral metagenomic sequencing to analyze the virome of fetal bovine serum (FBS) and trypsin collected from different vendors and source origins and update viral sequences either on current regulatory lists or not on our radar.
MATERIALS AND METHODS
In total, we collected 28 FBS and 25 trypsin samples from seven and five different vendors, respectively, in four main geographic regions. The FBS samples were produced in North America (NA,n=10), South America (SA,n=3), Australia (AU,n=10), and Asia (AS,n=5), and the trypsin samples were produced in North America (NA,n=10), Australia (AU,n=10),and Asia (AS,n=5) (Table 1). Samples from the same region were pooled together.
Sample processing, library construction, and sequencing
Viral metagenomic sequencing was performed as described previously (Li et al., 2019, 2021). In brief, the pooled FBS samples were centrifuged at 12 000 ×gfor 15 min at 4 °C, and the resulting supernatant was passed through a 0.45 μm sterile filter to reduce eukaryotic and bacterial material (Costar Spin-X centrifuge tube filters, Corning, USA). Filtrates were incubated for 90 min at 37 °C with a cocktail of nucleases to reduce the background of free nucleic acids. Viral nucleic acids were extracted using a Qiagen Viral RNA Mini Kit(Qiagen, Germany). To obtain enough material for library construction, nucleic acids were amplified using two randomamplification approaches: i.e., multiple displacement amplification (REPLI-g Single Cell WTA Kit, Qiagen,Germany) and random reverse-transcription polymerase chain reaction (RT-PCR) amplification as described previously (Li et al., 2020). The amplified nucleic acids were purified using a QIAquick PCR Purification Kit (Qiagen, Germany). Libraries were generated using a NEBNext Ultra II FS DNA Library Prep Kit (Illumina, USA), quantified using Qubit 3.0(Invitrogen, USA), and sequenced on the Illumina NovaSeq platform (Illumina, USA) at Novogene Co., Ltd. with 2×150 bppaired-end reads with dual barcoding for each sample pool.
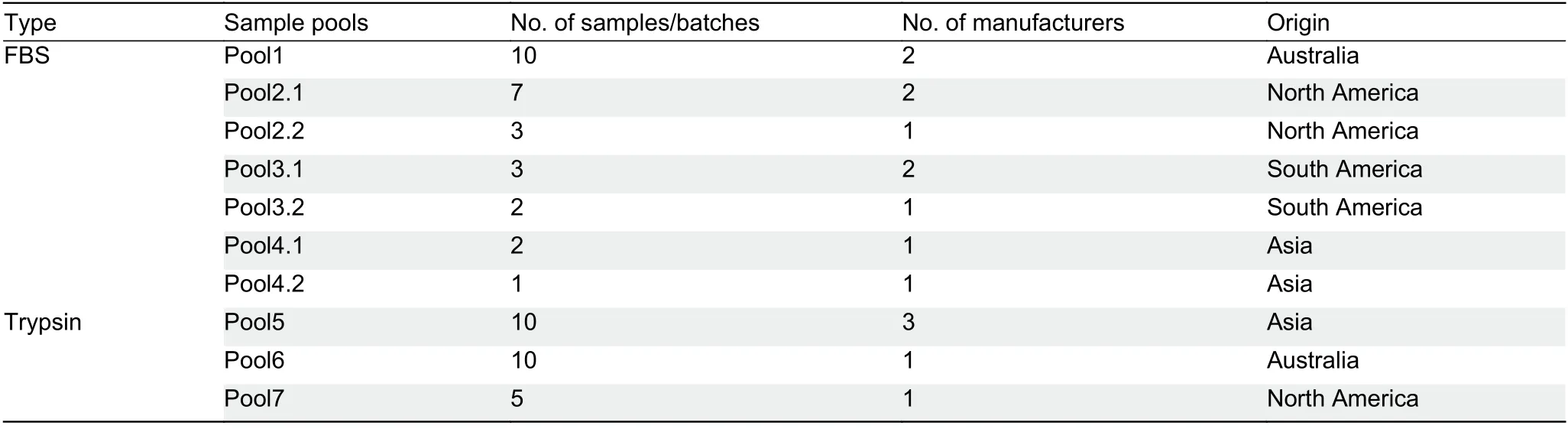
Table 1 Sample information for FBS and trypsin from 10 pools manufactured in four different regions
Bioinformatic analyses
Viral sequences were analyzed using an in-house pipeline as described previously (Li et al., 2022). Sequencing adaptors and low-quality sequences were removed using and Trimmomatic v.0.38 (Bolger et al., 2014). Host sequences were subtracted from the data using Bowtie2 v.2.3.4.3(Langmead & Salzberg, 2012). The remaining high-quality reads werede novoassembled using Megahit v.1.1.3 (Fu et al., 2012). Both singlets and assembled contigs were mapped against viral nucleic acid and protein databases selected from the NCBI nr/nt database (based on annotation taxonomy in Virus Kingdom) using BLASTn (E<10-10) and BLASTx (E<10-5) (DIAMOND v.0.9.24) (Buchfink et al., 2015),respectively. False positives (reads or contigs with higher similarity to non-viral than to viral sequences) were removed by mapping all potential viral hits against the NCBI nr/nt database (ftp://ftp.ncbi.nih.gov/blast/db). Viral reads and contigs were manually checked to exclude potential artifacts.Viral abundance was calculated by reads per million (RPM).
Phylogenetic analyses
Viral contigs/reads of each virus were extracted and aligned to reference viral genomes to generate full or partial genomes using the Geneious R11 program (Kearse et al., 2012). Viral nucleic acid sequences were first translated into amino acids,then aligned using MAFFT v7 (Katoh et al., 2019).Phylogenetic trees were inferenced using the maximumlikelihood method with MEGA X. The Models program in MEGA X was used to determine the best substitution model.Phylogenetic trees based on nucleotide sequences were generated using the bootstrap method (1 000 times) under a GTR+G+I model.
PCR detection of lyssavirus, Nipah virus, and anellovirus
Viral nucleic acids were extracted using a Qiagen Viral RNA Mini Kit (Qiagen, Germany). Reverse transcription was performed with a SuperScript III First-Strand Synthesis Kit(Thermo Fisher, USA). Universal primers from previous studies as well as primers designed based on assembled contigs were used for amplification of lyssavirus and Nipah virus. For lyssavirus, two sets of primers were used: JW12 5’-ATGTAACACCYCTACAATTG-3’, JW6 5’-CARTTVGCRCACA TYTTRTG-3’, 5’-GTCATYARWGTRTGRTGTTC-3’ (Heaton et al., 1997); F: 5’-AGAAAGCACTTCCGCCCTAC-3’, R1: 5’-CACCAGGTGCTCAATCTCGT-3’, R2: 5’-CATGTGCCGTCC ATCAGTCT-3’. For Nipah virus detection, two sets of primers were used: F1: 5’-TCITTCTTTAGAACITTYGGNCAYCC-3’,F2: 5’-GCCATATTTTGTGGAATAATHATHAAYGG-3’, R: 5’-CTCATTTTGTAIGTCATYTTNGCRAA-3’ (Tong et al., 2008);F1: 5’-CGTGGTTATCTTGAACCTATGTACTTCAG-3’, R1: 5’-CGCAACTTTAATGTAATTGGTCCCTTAGTG-3’, F2: 5’-CAG AGAAGCTAAATTTGCTGCAGGAGG-3’, R2: 5’-TCACAC ATCAGCTCTGACAAAGTCAAG-3’ (Yadav et al., 2019).Universal primers targeting the untranslated region (UTR) of anellovirus were used: F: 5’-ACWKMCGAATGGCTGAGTTT-3’, R: 5’-CCCKWGCCCGARTTGCCCCT-3’ (Thijssen et al.,2020). PCR consisted of 25 μL of Thermo Scientific DreamTaq Green PCR Master Mix (2×), 1.0 μmol/L Forward/Reverse primer, 3 μL of template DNA, and water to 50 μL. The PCR amplification procedures followed above studies or included: denaturation at 95 °C for 3 min, 40 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, with a final elongation step at 72 °C for 10 min. The PCR results were visualized on 1% agarose gel.
RESULTS
Overview of viral sequences
In total, we obtained 10 million paired-end reads, with a median of 2 233 723 sequences per sample pool. Followingde novoassembly, sequences were annotated using the virusonly nucleotide and protein databases (see Methods). Viral reads accounted for 1% of total reads (101 357 784), with 26%and 74% from eukaryotic and prokaryotic origins, respectively(Figure 1A). We identified sequences representing eight eukaryotic viral families.Parvoviridaewas the most abundant,followed byAnelloviridae,Flaviviridae,Herpesviridae,Caliciviridae,Nodaviridae,Rhabdoviridae, andParamyxoviridae(Figure 1B). In total, the genomes of eight different eukaryotic viruses were detected in FBS pools from North America, as well as four from Australia, six from South America, and six from Asia (Figure 1C). Details of these viruses, including read number, contigs, best-hit, identity, and genome coverage, are listed in Table 2. Viral genomes fromParvoviridaewere the most abundant sequences found in all sample pools (94.1% in North America, 99.9% in South America, 99.7% in Asia, and 56.7% in Australia), and bovine parvovirus 3 was the most abundant genome representative member of theParvoviridaefamily. TheParvoviridaegenomes also contained the most diverse virus species, including bovine parvovirus 3 (BPV3), bosavirus, bovine copiparvovirus 3 (BcoPV3), densovirus, and bovine hokovirus 2 (BHoV2).Compared to FBS, only three eukaryotic viruses were detected in trypsin pools from all three regions, possibly due to enzymatic activity or the inactivation and clearance of viruses by the host. Densovirus fromParvoviridaewas the most abundant viral genome in all trypsin samples (Figure 1C;Table 2). Reads mapped to lyssavirus were detected in trypsin from AU.
Parvoviridae
Parvoviridaeis a diverse family of single-stranded DNA viruses that can infect a variety of different mammals(Cotmore et al., 2019). As the most abundant family detected in the FBS and trypsin samples, viral sequences related to five different parvoviruses were found, i.e., BPV3, bosavirus,BcoPV3, densovirus, and BHoV2.
BPV3, which belongs to theErythroparvovirusgenus, was detected in 6/7 FBS pools, covering all four geographic regions. We successfully assembled six full-length or near fulllength BPV3 genomes (GenBank accession Nos.OM836168-OM836173). Phylogenetic analysis based on the non-structural (NS) and viral structural protein (VP) coding regions showed that all six BPV3 sequences clustered with several viruses previously found in China and Brazil(Figure 2). Among them, two BPV3 genomes from pools NA1 and AS shared more than 96% identity, with a closely related genome also identified in China (MG026727). Furthermore,both genomes shared ca. 90% identity to the other four BPV3 genomes (NA2, AU, SA1, and SA2), which clustered with a closely related BPV3 strain found in the US (AF406967).These data indicate that BPV3 is a common bovine-related virus in serum samples, and related BPV3 strains are circulating in different continents.
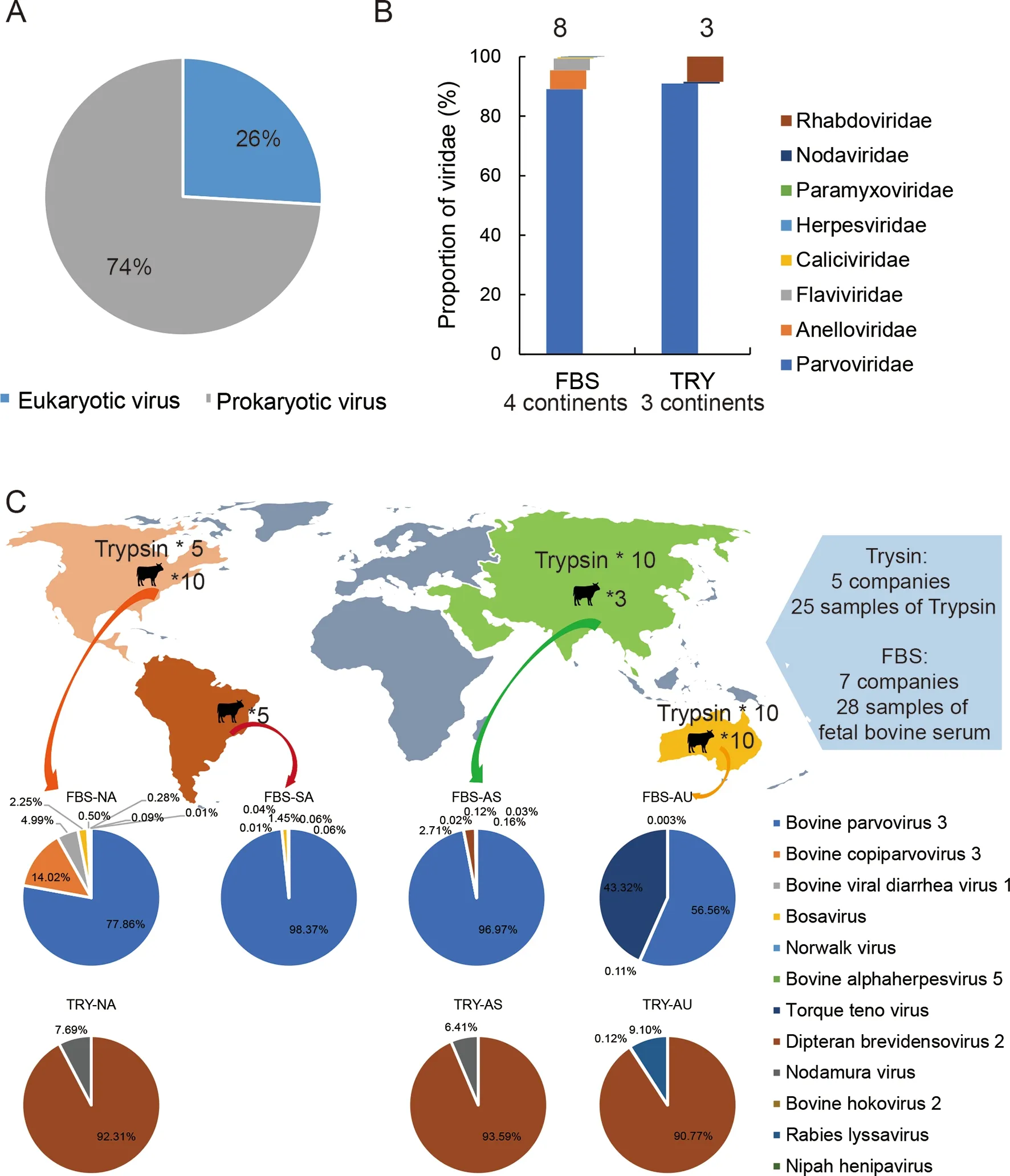
Figure 1 Profiles of all viruses in different sample pools and geographic regions
Bosavirus, which belongs to theCopiparvovirusgenus, was first discovered by metagenomic sequencing of calf serum from the US (Sadeghi et al., 2017); only two genomes have been reported so far. Here, bosavirus was detected in four FBS samples from North America, South America, and Australia. A complete genome of bosavirus (bovine bosavirus-NA, GenBank accession No. OM836179) was assembled from the FBS pools from North America, showing 96.9% and 96.8%identity in the NS and VP regions, respectively, to two viruses found in the US. In addition, partial bosavirus open reading frame (ORF) sequences were assembled from South America,accounting for 32.8% of the genome (Table 2). All bosavirus sequence reads and contigs showed high similarity(98.4%-99.2%) to viruses discovered in the US. These data indicate that bosavirus, a recently recognized virus in cattle,may be a common bovine virus circulating in different herds.
BcoPV3, another virus from theCopiparvovirusgenus, was first identified in pooled FBS samples from South America and Europe (Baylis et al., 2020). Here, we discovered more than 17 000 BcoPV3 reads in one North American FBS pool and assembled a complete genome (about 6 413 bp) (BcoPV3-NA,GenBank accession No. OM836180). BcoPV3 displayed high genetic identity in the NS and VP regions (98.6% and 98.7%,respectively) with the only two isolates sequenced to date(Figure 2).

Table 2 Summary of eukaryotic viral sequences retrieved in commercial batches of FBS and trypsin
Members ofDensovirinaeare highly divergent and currently classified into more than 10 genera. These viruses are known to infect invertebrates, mainly insects. Viral reads related toDipteran brevidensovirus 2were detected in 3/7 FBS pools and 3/3 trypsin pools. Densovirus contigs from North America FBS pool 1 showed the highest identity (92.6%) with a densovirus discovered from birds in China (MT138289). All other densovirus sequences (two from FBS and three from trypsin samples) were most closely related to a densovirus(AY310877) infecting mosquitos in Peru. Only partial densovirus genomes were recovered from three samples, and phylogenetic analysis showed that these partial genomes clustered with mosquito viruses (GenBank accession Nos.OM836174-OM836175).
BHoV has been previously detected in cattle from the US and China (Mitra et al., 2016; Xu et al., 2016). We only detected hokovirus reads in one FBS pool from South America, and all reads and assembled contigs from this pool were most closely related to the hokovirus (100% identity)discovered in the US.
Flaviviridae
BVDV, which belongs toPestivirusinFlaviviridae, contains a single-stranded positive RNA genome (9.7-15.7 kb). The virus is widespread, and most herds are at risk of infection. Here,BVDV was detected in FBS pools from North America, and its near full-length genome (about 15.1 kb) (BVDV-NA, GenBank accession Nos. OM836181 and OM836182) was successfully assembled. Phylogenetic analysis based on the near fulllength genome sequences showed that BVDV-NA belonged toPestivirus Aand shared only 87.2% identity with the closest relative found in China (Figure 3A). No BVDV sequences were found in any other sample.
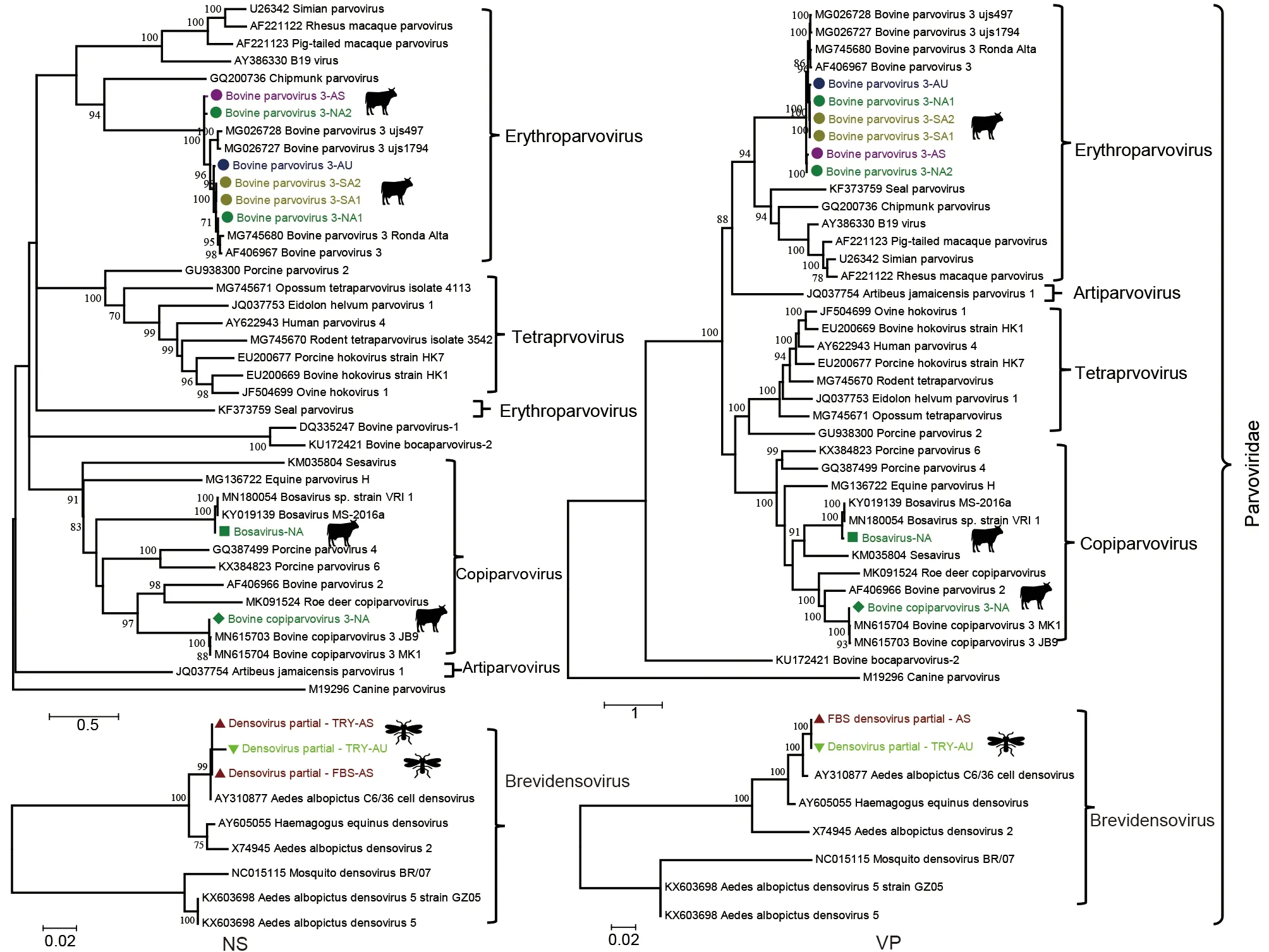
Figure 2 Evolutionary relationships of all parvoviruses found in this study with representative viruses from Parvoviridae
Caliciviridae
Noroviruses are RNA viruses belong to the genusNorovirus(familyCaliciviridae). Bovine noroviruses (BNoV) are frequently detected in fecal samples from diarrheic cattle (Di Felice et al., 2016; Khan & Alam, 2021). In our study, more than 1 300 reads were detected in a FBS sample pool from North America, and three contigs covering about 35% of the full genome were assembled (570 bp, 664 bp, and 949 bp in length, respectively) (GenBank accession Nos. OM817500,OM817465, and OM816743). Phylogenetic analysis was conducted using the largest contig sequence. Results showed that the partial genome belonged to the bovine-related norovirus GIII. Furthermore, the partial viral genome was most closely related to the norovirus GIII isolate BO/HN-1/2018/China. The two viral sequences shared 82.0%nucleotide identity, indicating a new genotype in FBS samples(Figure 3B).
Other viruses
Anelloviruses are highly prevalent and genetically diverse, and are present in most human populations as well as in some animals (Kaczorowska & Van Der Hoek, 2020; Shulman &Davidson, 2017). We detected anellovirus sequences in FBS pools NA1, AU, and AS1, respectively. Sequences from the three pools had the highest BLASTn hits to anelloviruses found in human serum (Table 2). The presence of anellovirus sequences in the three pools was confirmed by specific quantitative PCR. Bovine alphaherpesvirus 5, also known as bovine encephalitis herpesvirus, was detected in FBS pools NA1 and AS1. Sequence reads and contigs showed 93%-100% identity to BLASTn hits (KY559403) from Brazil.Nodamura virus, an RNA virus with segmented genomes, was found in three FBS pools and three trypsin pools. Although members inNodaviridaemainly infect insects and ticks,Nodamura virus is the only virus reported to infect insects,fish, and mammals (Johnson et al., 2003; Li et al., 2013). The Nodamura virus sequences found here showed high identity(98.8%-100.0%) to the closest hit in the database.
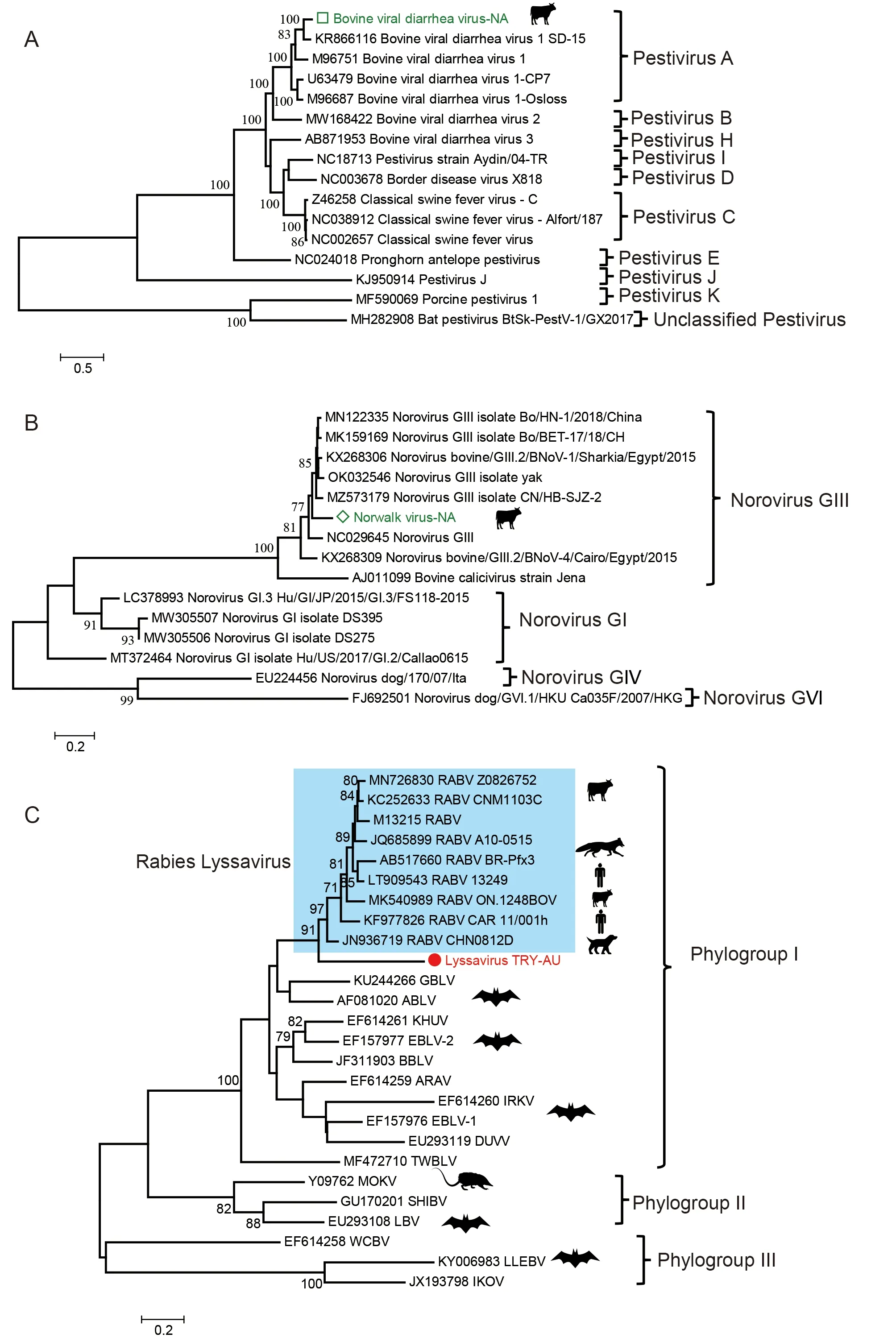
Figure 3 Evolutionary relationship of newly described BVDV, bovine norovirus, and lyssavirus
Viruses with unknown origin
We found the presence of viral reads belonging to two highly pathogenic human viruses, lyssavirus, and Nipah virus(Table 2). More than 140 lyssavirus reads were found in a trypsin sample from Australia. All sequences were mapped to the G gene (glycoprotein) of the virus. Phylogenetic analysis based on the largest contig sequence (454 bp) showed that the new sequence lay outside the rabies lyssavirus cluster and shared 76% identity with the most closely related rabies lyssavirus sequence (JN936719) and 55%-71% identity to all other representative lyssaviruses (Figure 3C). These results suggest that the partial sequences may be from a new lyssavirus species. We attempted to amplify the two viruses using universal nested RT-PCR targeting a conserved region of lyssavirus (Heaton et al., 1997) and specific primers designed based on the assembled contigs, but both methods failed to amplify the virus from both the original samples and random amplification products. We also detected 34-72 Nipah virus reads in two FBS samples from Asia and South America.Both viral reads and contigs showed high identity(99%-100%) to other Nipah viruses in the database. Same as the lyssavirus, nested RT-PCR failed to amply the viral sequences from the samples.
To trace the potential origin of the two viruses, we analyzed all sequencing data generated from previous and subsequent libraries in our laboratory, but found no viral sequences related to either virus. Furthermore, no studies related to either virus have been conducted in our facility, thereby excluding possible contamination from our lab and facility. However, we cannot exclude the rare possibility that they may be contaminations from other samples on the sequencer of the company, which accidentally contained potential new lyssavirus and Nipah virus sequences.
DISCUSSION
Animal-derived biologicals are important supplements for basic and medical research and pharmaceutical production(Paim et al., 2021; Van Der Valk et al., 2018). Although certain regulations (USDA and EMA) and practices are followed by manufacturers, most methods are based on traditional virological assays, including virus isolation and immunofluorescence, which typically target known animal viruses. Continuous detection of animal viruses, including novel and human-related viruses, from biological samples demonstrates the limitations of current safety guidelines(Baylis et al., 2020; Cheval et al., 2011; Gagnieur et al., 2014;Paim et al., 2021; Paim et al., 2019; Sadeghi et al., 2017).
In this study, we collected FBS and trypsin samples from different vendors and geographical locations and characterized their viromes using unbiased viral metagenomics. Results showed that the samples were not virus-free, with at least two viruses detected in each sample pool. Viral reads fromParvoviridaewere the most abundant,followed byAnelloviridae. Due to their non-enveloped nature and small size, viruses from these two families may be less sensitive to filtration and irradiation measures (Pecora et al.,2020; Sadeghi et al., 2017), which may explain their high prevalence. We detected complex diversity of bovine-related viruses, including BPV3, BcoPV3, bosavirus, and BHoV2 fromParvoviridae, BVDV1 fromFlaviviridae, BNoV fromCaliciviridae, and bovine alphaherpesvirus 5 fromHerpesviridae. Most of these bovine-related viruses are associated with diarrhea, encephalitis, and respiratory disease in cattle (Barnes et al., 1982; Del Médico Zajac et al., 2010;Gomez-Romero et al., 2021; Jung et al., 2014). Even though centrifugation, filtration, and nuclease digestion were performed during sample pretreatment, the detection of viral sequences does not necessarily imply pure viral particles or active viruses in the sample as they could be from residual free nucleic acids. However, their presence in commercial serum and trypsin samples, including several animal viruses already on the current detection list (Marcus-Sekura et al.,2011), highlights the need for stricter regulations during the production of biologicals. For example, the use of metagenomic sequencing could greatly increase the ability to detect a wide range of viruses as well as monitor emerging ones (Cantalupo & Pipas, 2019; Chiu & Miller, 2019; Harvey &Holmes, 2022).
The high prevalence and abundance of BPV3 and bosavirus in the different FBS pools indicated a high burden of these bovine viruses in different regions. Several other parvoviruses were also detected, albeit less frequently. These results suggest that current preventive measures may not be effective against parvoviruses, and the inclusion of additional parvoviruses, especially BPV3 and bosavirus, in routine testing is required. Due to extensive international trade,viruses from one affected farm or factory can be detected in different countries/regions where contaminated FBS products have been used. For example, BHoV2 sequences in South American FBS were identical to a virus previously found in feedlot cattle in the US (Mitra et al., 2016). Commercialized bovine serum is usually produced by pooling thousands of individual samples, and product origin is usually labeled with the region where processing and packaging occurred (Paim et al., 2021). Thus, tracing viral origin can be challenging.
Animals often live alongside mosquitos and ticks, which are important factors in vector-borne viruses. Therefore, herds may carry insect-related viruses through bites (Schneider et al., 2021; Vonesch et al., 2019). The high prevalence of mosquito-related densovirus and Nodamura virus among FBS and trypsin pools suggests considerable interactions between herds and insects. Although insects are the most common hosts of Nodamura virus, several studies indicate that the virus can also infect mammals (Johnson et al., 2003; Li et al.,2013). Currently, however, little is known about the presence and prevalence of densovirus and Nodamura virus in farm animals and whether these viruses can infect humans.
We detected anellovirus in three FBS sample pools, with the most closely related anellovirus previously detected from human blood. Of concern, human viruses may be inadvertently introduced into biologicals during sample collection and production processes (Paim et al., 2021).Anelloviruses are widespread without apparent pathogenicity.However, for pathogenic human viruses, contamination in these biologicals may pose a higher potential risk, especially if viruses are not fully inactivated during production. Thus,stricter sterilization as well as global detection methods targeting both animal and human viruses should be considered in the future.
Lyssavirus and Nipah virus are highly pathogenic zoonotic viruses naturally found in bats (Kohl & Kurth, 2014; Van Brussel & Holmes, 2022). Spillover of both viruses has been reported in humans and domestic animals, including dogs,horses, cats, cattle, goats, sheep, and pigs (Benavides et al.,2020; Chao et al., 2021; Shipley et al., 2019; Siepker et al.,2020). Pigs are the most common intermediate host of Nipah virus, but other domestic animals such as cattle, horses,goats, and dogs can also be infected (Chowdhury et al., 2014;Field & Kung, 2011; Mills et al., 2009). Here, both viruses were detected in FBS and trypsin samples by metagenomic sequencing, although not confirmed by the RT-PCR, which may be due to low viral loads and/or degradation of the samples. Other possibilities for their presence may be contamination from the sequencing company. For example,modified rabies virus is commonly used as a vaccine vector targeting different viruses, including Nipah virus (Chi et al.,2022; Keshwara et al., 2019; Kurup et al., 2015), and the sequencing of materials carrying these artificial constructs may lead to accidental contamination in FBS and trypsin samples. However, the high genetic distance of the new lyssavirus sequence to any known sequence in the database raises the possibility that an emerging bat-origin lyssavirus is circulating in farm animals.
The detection of complex viruses in FBS and trypsin samples indicates the need to improve the safety and quality of these biologicals. However, the detection of viral genome sequences does not necessarily imply the presence of viral particles and/or active viruses, as these viruses could be attenuated or inactivated by irradiation during production,presence of neutralizing antibodies in serum, and/or long-time storage. Therefore, virus isolation is required to determine whether live viruses are present in FBS and trypsin samples.
In addition to the potential safety risks of animal and human virus contamination, whether large quantities of viral genomic elements can lead to unexpected or spurious results during cell culture experiments also needs to be addressed. For example, intake of viral nucleic acids may be recognized by the cellular innate immune system (e.g., Toll-like receptors(TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors(RLRs), and/or NOD-like receptors (NLRs), thereby triggering downstream signaling pathways (Brubaker et al., 2015; Cai et al., 2021). Unexpected gene expression may affect cell growth status or produce false results due to additional stimulation.
In summary, we detected viral sequences belonging toParvoviridae,Flaviviridae,Herpesviridae, andCaliciviridaein FBS and trypsin samples from different geographic regions.The presence of different animal viruses in such products highlights the need for more robust detection measures for quality control and monitoring of emerging viruses with implications for animal and human health.
DATA AVAILABILITY
Raw sequencing data were deposited in the following databases: Genome Sequence Archive (GSA, https://ngdc.cncb.ac.cn/gsa/), project No. PRJCA010138, accession Nos.SAMC806880-SAMC806883 and SAMC802758-SAMC802 763; Science Data Bank (https://www.scidb.cn/en), CSTR:31253.11.sciencedb.j00139.00015, DOI:10.57760/sciencedb.j00139.00015; CNSA (https://db.cngb.org/cnsa/) of CNGBdb,project No. CNP0002738, accession Nos. CNS0523595-CNS0523604. Viral sequences generated in this study can be found at GenBank (GenBank accession Nos. OM836168-OM836182, OM816743, OM817465, OM817500, and ON695913).
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Y.P.L. and C.Y.Z. participated in study design; P.Z. and Y.Y.M. collected the samples; P.Z. and Y.Y.M. performed the main experiments; P.Z., Y.Y.M., Y.P.L., and L.C. performed data analyses; P.Z., C.Y.Z., and Y.P.L. interpreted the data;Y.P.L. wrote the manuscript. C.Y.Z. and B.S. contributed to data interpretation and manuscript revision. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We would like to thank Prof. Jian-Hua Wang (Guangzhou Institutes of Biomedicine and Health, CAS) and Prof. Yi-Qun Kuang (First Affiliated Hospital of Kunming Medical University,Kunming Medical University) for their support in manuscript discussion and revision. We also thank Ms. Rui Yu at the Shanghai Public Health Clinical Center for help in sample collection.

- Zoological Research的其它文章
- Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing
- Coevolutionary insights between promoters and transcription factors in the plant and animal kingdoms
- Deficiency of transmembrane AMPA receptor regulatory protein γ-8 leads to attention-deficit hyperactivity disorder-like behavior in mice
- Global cold-chain related SARS-CoV-2 transmission identified by pandemic-scale phylogenomics
- The Hippo pathway and its correlation with acute kidney injury
- Genomics and morphometrics reveal the adaptive evolution of pikas