
STAT3与环烷烃并噻吩类衍生物抑制剂的分子对接
2022-10-15 09:02:46高鸿雁史丽颖曹洪玉于大永
天津师范大学学报(自然科学版) 2022年5期
高鸿雁,史丽颖,宋 昱,曹洪玉,于大永
(大连大学 生命科学与技术学院,辽宁 大连 116622)
信号传导和转录激活因子(signal transducer and activator of transcription,STAT)家族包含7个成员:STAT1、STAT2、STAT3、STAT4、STAT5a、STAT5b和STAT6[1].STAT蛋白有5个结构域,即氨基末端结构域、卷曲螺旋结构域、DNA结合结构域、SH2结构域和C端反式激活结构域[2].STAT1、STAT3、STAT5在癌细胞的增殖、侵袭和转移中起着重要作用,其中STAT3最为活跃[3].STAT3是肿瘤免疫治疗的一个热门靶点,STAT3抑制剂不仅可以直接作用于肿瘤细胞抑制其生长和转移,还可以调节肿瘤的微环境如炎症、血管新生和肿瘤免疫等[4],开发新型的特异性STAT3小分子抑制剂用来治疗癌症一直是学术界的热点问题之一.
目前已报道的STAT3抑制剂有很多,其中作用机制清楚的STAT3抑制剂主要包括1,4-萘醌类[5]、喹啉类[6]、苯并[b]噻吩-1,1-二氧化物类衍生物[7]、氯硝柳胺衍生物[8]、水杨酸衍生物[9]等.已有多个STAT3抑制剂小分子进入临床实验,如氯硝柳胺、乙胺嘧啶、C188-9、BP-1-102等[10-11].SH2区域是STAT3蛋白中最为保守的一个结构域,对STAT3磷酸化二聚体的形成至关重要,直接作用于STAT3蛋白的SH2区域可阻断STAT3的磷酸化、二聚化、核转移以及STAT3靶基因的表达.周文波[12]合成了一系列靶向STAT3的SH2结构域的2-氨基-3-氰基噻吩衍生物类小分子抑制剂,将STAT3小分子抑制剂的活性提高了10倍左右.
本课题组前期对2-氨基-3-氰基噻吩衍生物类小分子进行了三维定量构效关系的研究[13],在此基础上,本研究利用分子对接技术进一步阐明了2-氨基-3-氰基噻吩衍生物类小分子与STAT3的SH2结构域相结合的作用方式,为新型的靶向STAT3小分子抑制剂的设计开发提供了理论依据.
1 材料与方法
1.1 蛋白及小分子处理
从蛋白质晶体结构数据库PDB(http://www.rcsb.org/)搜索下载STAT3蛋白晶体结构,PDB编号为6HJS.利 用Schrödinger 2017软 件 包 中 的Protein Preparation Wizard工具对蛋白结构进行加氢、去除水分子、补全不完整残基、删除多余蛋白构象、分配相关电荷和能量最小化处理.从文献[12]中获取52个2-氨基-3-氰基噻吩衍生物类小分子的化合物结构,如图1所示,通过ChemOffice软件进行结构绘制并转换为3D结构.将小分子的3D结构导入Schrödinger 2017软件包中,利用Ligprep工具添加OPLC_2005立场进行结构优化,最终得到能量最低的分子构象,将其保存为.mol2格式.使用AutoDock和AutoDock Vina进行分子对接时,借助OpenBabel程序将小分子的.mol2格式转换为.pdbqt格式.

图1 52个小分子化合物的结构Fig.1 Structure of the 52 small molecules
1.2 计算程序筛选
为了获得准确可靠的分子对接结果,分别运用AutoDock、AutoDock Vina[14]、InsightⅡ中的CDOCKER(精准分子对接)程序以及Schrödinger 2017软件中的Glide XP(精确对接)4种对接手段对STAT3与其共晶化合物进行对接,根据对接后的小分子与原配体分子的均方根偏差(root-mean-square deviation,RMSD)的大小,判断对接程序与对接参数设置的合理性以及对该晶体复合物的适用性,一般认为RMSD≤0.2 nm时能够较好地重现原配体-受体的结合模式.最终筛选出最适合本研究的分子对接程序及方法.
1.3 分子对接
本研究中STAT3晶体结构中的共晶化合物结合于STAT3的SH2结构域,因此采用配体扩张法确定活性区域,即以原配体位置为中心向外扩张一定的范围生成对接盒子(docking box),此范围内的受体残基就构成了相关的活性位点,对接盒子的大小会影响对接结果的准确性和可靠性.对接盒子的中心设置为X=13.24,Y=54.43,Z=0.27,AutoDock中对接盒子大小设为5.6 nm×8.2 nm×6.2 nm,AutoDock Vina中设为2.06 nm×3.11 nm×2.31 nm,CDOCKER中对接盒子设为半径2.0 nm的球体,Glide XP中对接盒子大小设为2.3 nm×3.1 nm×2.1 nm,图2为Glide XP中生成的活性区域盒子.借助OpenBabel程序将AutoDock与AutoDock Vina产生的对接结果文件转换为.pdb格式并导入Schrödinger 2017软件中,借助Pose viewer模块分析对接构象以及蛋白-小分子间的相互作用方式.Glide XP中,利用Ligand Docking工具对靶蛋白与小分子进行对接,利用Ligand Docking模块中的对接分数(docking score)打分函数值用来评价小分子与靶蛋白的相互作用.
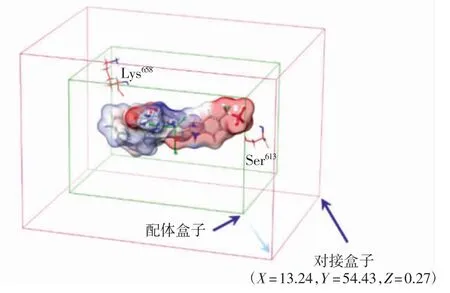
图2 配体扩张法生成的对接盒子Fig.2 Docking box generated by the ligand expansion method
2 结果与分析
2.1 计算程序筛选
本研究考察了AutoDock、AutoDockVina、CDOCKER和Glide XP这4种分子对接程序对STAT3与其原配体对接的重现性,原配体构象与对接后构象的叠合图如图3所示,4种对接程序的RMSD值均小于0.2 nm,如表1所示.
由表1和图3可知,使用4种程序进行分子对接均具有良好的准确度和可靠性,其中,Glide XP与AutoDock Vina的RMSD较低,Glide XP对接结果中配体分子在与STAT3蛋白的结合方式上与原配体更加类似,而且可提供更详尽的对接信息,因此,后续选用Glide XP进行分子对接研究.

图3 原配体构象与对接后构象的叠合Fig.3 Superposition of the original ligand conformation and the docked conformation

表1 不同程序的分子对接结果Tab.1 Molecular docking results using different programm
2.2 分子对接
采用Glide XP程序对STAT3蛋白与52个2-氨基-3-氰基噻吩衍生物小分子进行分子对接,52个小分子的对接分数如图4所示.由图4可知,所有小分子的对接分数分布在-31.4~-22.6 kJ/mol之间,说明这些小分子均能较好地结合于STAT3蛋白的SH2结构域,其中,小分子52#的对接分数最低,为-31.4 kJ/mol,说明该分子的对接效果最好.
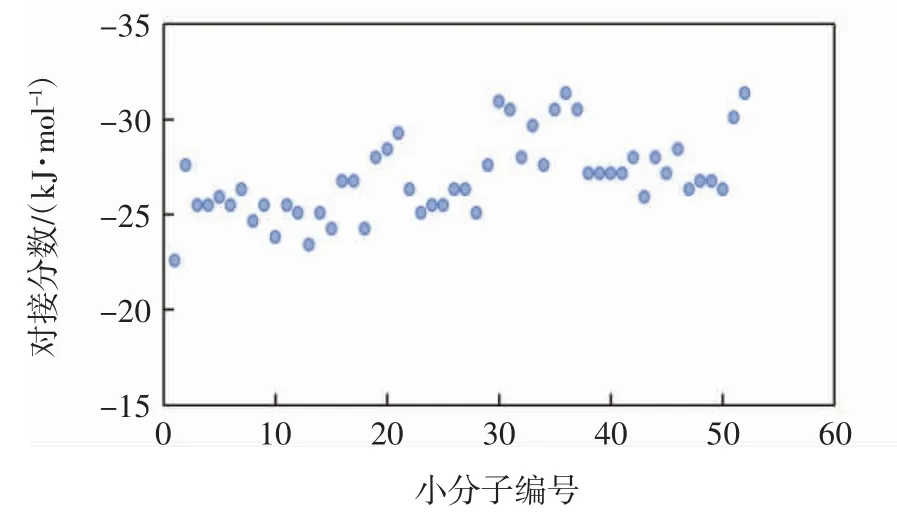
图4 52个小分子的Glide XP对接分数Fig.4 Glide XP docking scores of 52 small molecules
小分子52#能较好地进入蛋白表面的活性空腔,但与原配体相比未能进入侧面空腔,如图5所示,这也导致了小分子52#的对接分数差于原配体的对接分数.这与小分子结构的形状和大小有关,因此在后续的药物分子设计中,对小分子52#的构象进行合理改造,形成“桥梁”状结构也许有利于抑制剂活性的提升.
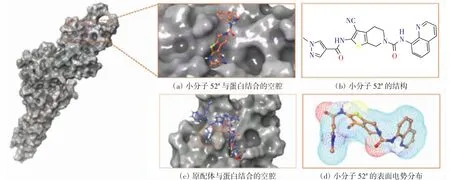
图5 小分子52#和原配体与STAT3结合的蛋白空腔以及小分子52#的表面电势分布Fig.5 Protein cavity of small molecule 52#and protoligand separatelybinding to STAT3 protein and the surface potential distribution diagram of small molecule 52#
2.3 互作模式分析
配体与受体的能量匹配包括静电作用、氢键作用、范德华相互作用和疏水作用等,其中,静电作用、氢键作用和范德华力控制药物-受体结合,疏水作用是药物-受体结合的驱动力.分析52个活性小分子与STAT3靶标SH2结构域的相互作用方式,发现氢键、极性疏水和静电作用力为主要的作用方式.对接分数排名前4位的小分子(52#、36#、31#、35#)与STAT3蛋白之间的互作模式如图6所示,其中,小分子52#与氨基酸 残 基Ser611、Glu612、Ser613形 成 氢 键 作 用,与Val637、Pro639、Trp623、Tyr657形 成 疏 水 作 用,与Lys591形成π-阳离子作用(π-cation),与Arg609、Glu612、Glu638、Lys591形成静电作用,与Ser611、Ser613、Thr620、Ser636形成极性作用.
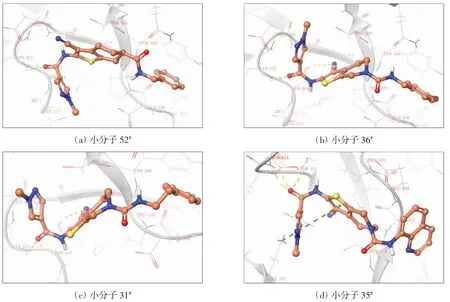
图6 对接分数排名前4位的小分子与STAT3蛋白之间的相互作用方式Fig.6 Interaction between STAT3 protein and small molecules in the top 4 docking scores
为了进一步考察2-氨基-3-氰基噻吩衍生物抑制剂与STAT3蛋白SH2结构域结合的关键氨基酸残基,对52个活性分子与蛋白结合的每一种作用类型所涉及的氨基酸残基进行分析.总结SH2结构域中氨基酸残基与52个小分子发生相互作用的数目,结果如图7所示;每一种相互作用类型中,能够与某一氨基酸残基发生作用的小分子数目如图8所示.由图7可知,Lys591、Arg609、Ser611、Glu612、Ser613这5个氨基酸参与的相互作用均在60次以上.由图8可知,52个小分子与氨基酸Pro639均形成疏水作用,与氨基酸Glu638均形成静电作用,只有1个分子没有与氨基酸Val637形成疏水作用.另外,40个以上的小分子与氨基酸Lys591、Arg609、Glu612形成静电作用,与Ser611、Ser613形成极性作用.
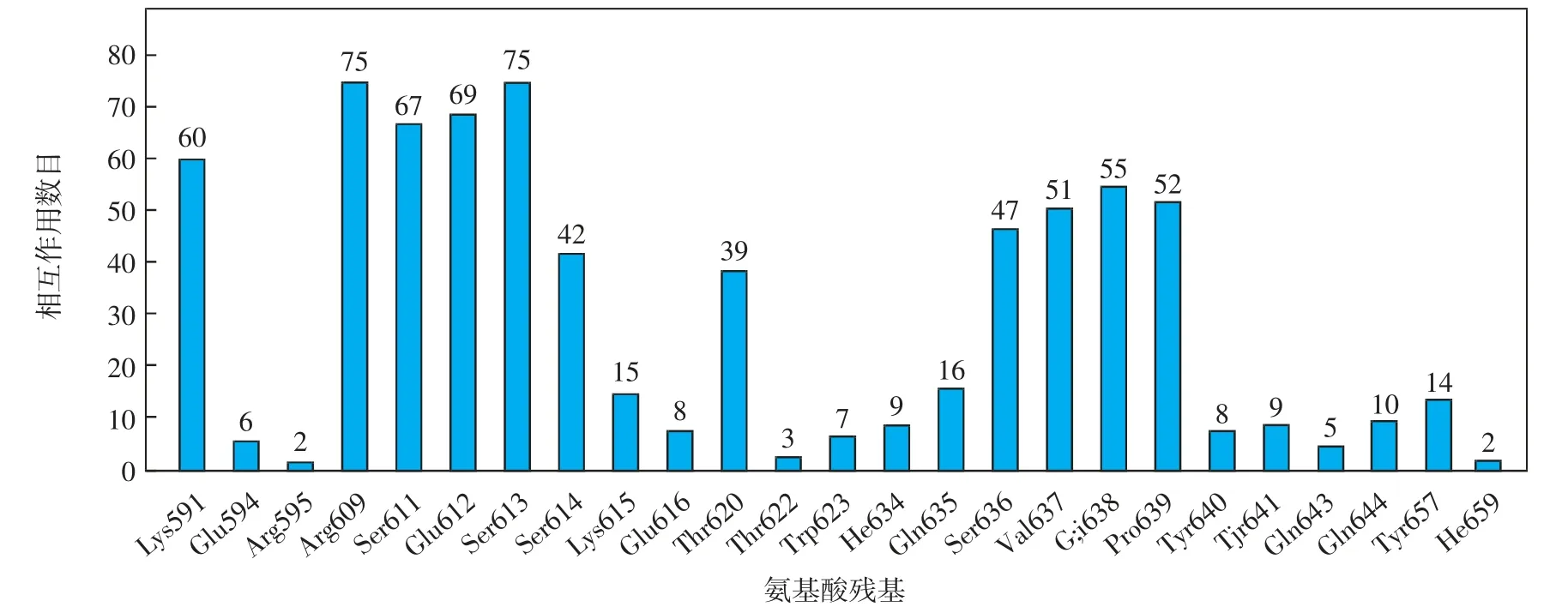
图7 SH2结构域的活性区域中氨基酸残基与小分子发生相互作用的数目Fig.7 Number of interactions between amino acid residues and small molecules in the active region of the SH2 domain
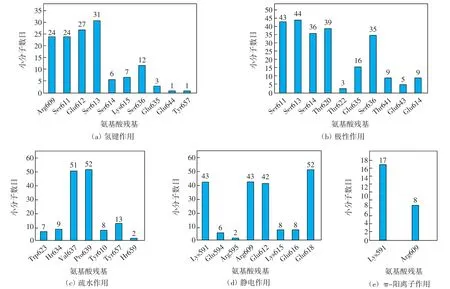
图8 不同作用类型中与氨基酸残基发生作用的小分子数目Fig.8 Number of small molecules interacting with amino acid residues in different types of interaction
综上所述,Lys591、Arg609、Ser611、Glu612、Ser613、Val637、Glu638、Pro639这8个氨基酸为2-氨基-3-氰基噻吩类衍生物与STAT3蛋白SH2结构域相互作用的关键氨基酸.在52个小分子化合物中,具有良好的STAT3抑制活性的化合物结构特征为:能够与Arg609、Glu612、Ser613形成氢键作用,与Val637、Pro639形成疏水作用,与Lys591、Arg609、Glu612、Glu638形成静电作用,与Ser611、Ser613形成极性作用.
3 讨论与结论
STAT3是细胞增殖、存活和凋亡的关键调节剂,在大多数人类癌症中被组成性激活[15],可以成为癌症治疗的重要潜在治疗靶标[16].目前临床上的STAT3小分子抑制剂的活性大多处于微摩尔级,因此,开发新型高效的特异性STAT3小分子抑制剂至关重要.本研究以文献[12]报道的52个新型的2-氨基-3-氰基噻吩衍生物类STAT3靶向抑制剂为研究对象,基于分子对接技术探究了该类抑制剂与STAT3蛋白SH2结构域的相互结合模式.结果表明,2-氨基-3-氰基噻吩衍生物类抑制剂与STAT3有良好的亲和力,氢键、疏水、π-cation、静电以及极性作用为主要的相互作用方式.通过对小分子-受体结合区域涉及相互作用的氨基酸残基进行分析发现,Lys591、Arg609、Ser611、Glu612、Ser613、Val637、Glu638、Pro639这8个氨基酸为2-氨基-3-氰基噻吩类衍生物与STAT3蛋白SH2结构域相互作用的关键氨基酸.另外,与STAT3晶体结构中的原配体相比,对接效果最好的小分子52#虽然能较好地进入蛋白表面的活性空腔,但受分子大小与形状的影响,未能进入侧面空腔.因此,在未来对于新型2-氨基-3-氰基噻吩衍生物类STAT3靶向抑制剂进行药物设计时,增加分子大小使小分子形成“桥梁”状结构也许有利于活性的提升.与临床药物相比,2-氨基-3-氰基噻吩衍生物类小分子抑制剂虽然活性提高了10倍左右,但仍有很大的提升空间,后续可以借助计算机辅助药物的手段设计新型的STAT3靶向小分子抑制剂并进行生物活性验证,期望得到高效的特异性STAT3小分子抑制剂.
猜你喜欢
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
四川警察学院学报(2019年6期)2019-12-28 07:20:06
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
文化产业(2016年6期)2016-10-19 19:13:47
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
合成化学(2015年10期)2016-01-17 08:56:26
应用化工(2014年9期)2014-08-10 14:05:08
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
无机化学学报(2014年10期)2014-02-28 17:33:13
