
杨梅素糖苷衍生物的合成及其抗菌活性研究
2022-10-14 07:02吴光旭吴红林李吉顺李天磊2吴松潘卫东
中国抗生素杂志 2022年8期
吴光旭 吴红林 李吉顺 李天磊2, 吴松,* 潘卫东
(1 贵州医科大学,药学院/省部共建药用植物功效与利用国家重点实验室,贵阳 550025;2 贵州省中国科学院天然产物化学重点实验室,贵阳 550014;3 中国医学科学院&北京协和医学院药物研究所 天然产物活性物质与功能国家重点实验室,北京 100050)
黄酮类天然产物广泛分布于植物界中,该类天然产物具有广泛的生物活性,如抗菌、抗炎、抗肿瘤、抗氧化、提高免疫力等,不同类型的黄酮类化合物被广泛应用于医药、食品、保健品、化妆品等领域。近年来,黄酮类化合物的抗菌活性日益受到重视[1],黄酮类化合物主要通过抑制细菌DNA合成[2]、改变膜电位和细胞膜功能[3]及抑制细菌能量代谢[4-5]等多种作用机制发挥抗菌作用。前期研究显示不同植物来源的黄酮提取物具有较为广泛的抗菌活性[6],尤其是3-羟基黄酮类天然产物活性较为突出,其主要的抗菌活性-构效关系总结如图1。目前,针对槲皮素(quercetin)、高良姜素(galangin)、芦丁(rutin)、山柰酚(kaemperol)及其结构修饰产物等的抗菌活性研究较多(图1)[7-10],但是对杨梅素(myricetin)及其糖苷衍生物抗菌活性研究较少。

图1 具有抗菌活性的代表黄酮苷类衍生物及其构效关系总结[7-10]Fig.1 Summary of representative flavonoid glycoside derivatives and structure-activity relationship[7-10]
当前,细菌对抗菌药物的耐药性日益增强,正成为对公共健康的严重威胁。在临床细菌感染疾病中,几乎70%的病原菌表现出对一种或多种抗菌药不同程度的耐受[11-12]。在当前细菌对临床药物耐受全面加剧和新型抗生素研发进展缓慢的双重夹击下,从天然产物中寻找和发现抗菌先导化合物是当下发现新型抗菌药物最具前景的途径之一[13-15]。为了发现具有抗菌活性的黄酮醇类化合物,本研究以天然产物杨梅苷作为原料,通过不同的保护策略、烷基化、酯化反应和糖苷化反应制备合成了14个杨梅素衍生物,对目标化合物进行了金黄色葡萄球菌、肠球菌和表皮葡萄球菌抑菌活性测试,并对其抗菌活性构效关系进行了初步探索。
1 材料
1.1 主要仪器
磁力搅拌器 (IKA RCT Basic),旋转蒸发仪(EYELA SB-1300),超导核磁共振波谱仪(Bruker AV-500),高效液相-单级杆质谱联用仪(Waters ACQUITY QDa),高效液相色谱仪(Shimadzu LC-6AD),低温反应仪器(EYELA PSL-1810),酶标仪(Multiskan FC) 。
1.2 主要试剂
N,N-二甲基甲酰胺,甲醇,四氢呋喃,异丙醇,盐酸-二氧六环溶液,碳酸铯,4-二甲氨基吡啶,二环己基碳二亚胺,钯碳,溴苄,对甲苯磺酸-一水合物,全乙酰化半乳糖,杨梅苷。
2 实验方法
2.1 杨梅素类衍生物合成
前期文献报道,3-羟基黄酮醇类半乳糖苷类衍生物显示较好的抗菌活性,其中山奈酚糖苷类化合物(1,图1),经过结构优化,在苷元3-羟基黄酮醇B环上引入羟基,同时用半乳糖修饰得到了对耐甲氧西林金黄色葡萄球菌抗菌活性更好的先导化合物(2,图1)[2]。基于前期研究结果,为了进一步探索3-羟基黄酮醇类抗菌活性-构效关系,设计合成了3-羟基黄酮醇B环三羟基取代半乳糖苷衍生物。由于多取代黄酮醇类化合物的制备方法受到取代羟基的影响较大,为了快速高效获取一定量的化合物用于活性筛选,采用了以天然杨梅苷作为原料进行选择性糖基化修饰策略。首先,利用天然杨梅苷(B环具有3个羟基取代)(4,图2)作为原料,通过杨梅苷3-位糖基残基可以对3-位形成天然的保护优势,经过苄基保护和直接酸解的方法脱除糖基残基得到了3-位羟基游离的黄酮醇中间体5。然后,中间体5与α-溴代-2,3,4,6-四乙酰半乳糖苷糖苷反应制备得到全乙酰化β-半乳糖苷,紧接着在碱性条件下脱除乙酰基得到中间体6。在酸性条件下,中间体6通过选择性丙酮叉基保护半乳糖3′′,4′-二羟基,该中间体7可以直接通过酰化反应对半乳糖2′,6′′-二羟基进行修饰,制备糖基选择性修饰的化合物。最后,针对半乳糖2′′,6′′-二羟基修饰官能团的耐受性,通过不同的脱苄基保护策略和羟基脱保护制备相应的目标产物9和10,具体合成路线及反应条件如图2所示。同时,中间体5的3-位游离羟基通过与不同的酰基试剂反应,可以高效制备3-羟基酰化产物11,其制备方法和结构表征参考已报道方法[16]。
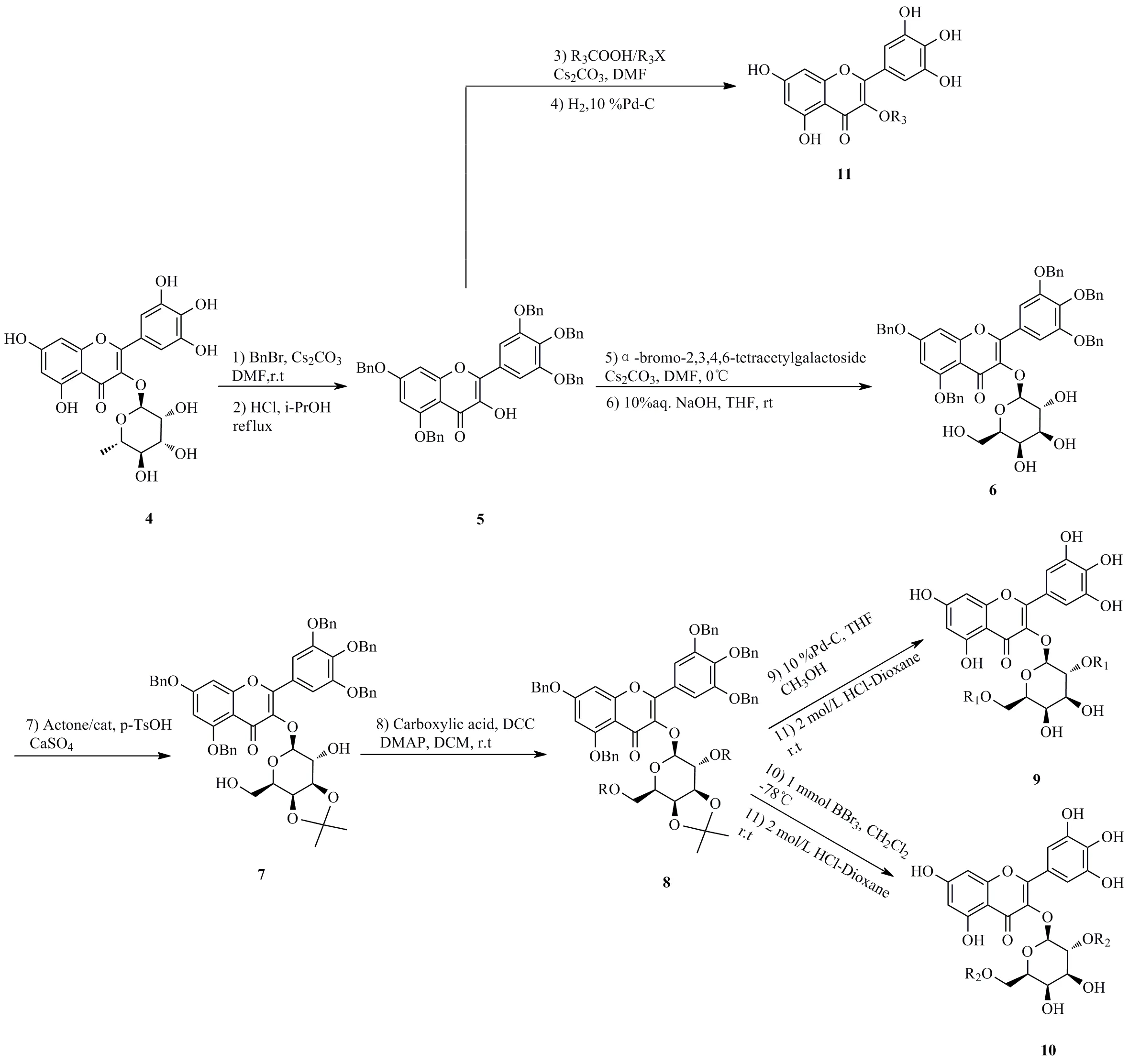
图2 杨梅素糖苷类衍生物的合成路线Fig.2 Synthetic route of myricetin glycoside derivatives
通过以上方法,共计合成新型杨梅素糖苷化合物8个:9a-1~9a-5和10a-1~10a-3,其中在半乳糖2′′,6′′-位进行了不同取代的苯丙酰基和肉桂酰基修饰;另外还有6个3-位羟基不同酰化修饰的杨梅素衍生物11a-1~11a-6。目标产物结构式如表1所示。

表1 合成的杨梅素糖苷类衍生物Tab.1 Synthetic myricetin glycoside derivatives
中间体5:5,7-双(苄氧基)-3-羟基-2-(3,4,5-三(苄氧基)苯基)-4H-苯并吡喃酮
在反应瓶中加入杨梅苷37.1 g(80 mmol),Cs2CO3156.5 g(480 mmol)及DMF(200 mL),0.5 h内滴加溴苄58.0 mL(480 mmol)。氩气保护下,室温搅拌过夜。用EtOAc(300 mL)稀释反应液后过滤,EtOAc(2×50 mL)洗涤、滤液用饱和氯化钠洗(3×100 mL)、无水硫酸钠干燥,过滤、减压旋干得到红棕色黏稠物。将上述红棕色黏稠物溶解在i-PrOH(300 mL)中,加入HCl(3 mL),加热回流反应15 min后,有沉淀析出。继续搅拌至原料反应完毕(LC-MS监测),过滤、EtOH(2×50 mL)洗涤,以EtOAc重结晶、干燥即得中间体5 46.7 g,淡黄色固体,收率为76%。m.p.172℃~174℃,ESI-MSm/z: 769.36 [M+H]+,C50H40O8,WM=768.27;1H NMR(400 MHz,DMSO-d6)δ 9.34 (s,1H),7.92-7.16 (m,27H),6.98 (s,1H),6.72 (s,1H),5.27 (s,4H),5.20 (s,4H),5.04 (s,2H)。
中间体6:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-半乳糖基-4H-苯并吡喃酮
在反应瓶中加入15.4 g中间体5(20 mmol),加入200 mL干燥DMF,搅拌溶解后,加入9.8 g(30 mmol)Cs2CO3,0℃下搅拌反应10 min后,加入12.3 g(30 mmol)新制备的α-溴代-2,3,4,6-四乙酰基半乳糖苷,0℃下继续搅拌过夜,次日LC-MS监测至反应完全,将反应液倒入500 mL冰水中,加入500 mLEtOAc萃取,然后以EtOAc(2×200 mL)再次萃取、合并有机相,水洗(3×500 mL)后,饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压旋干得淡黄色黏稠物,加入200 mL THF溶解上述黏稠物,加入200 mL 10%NaOH溶液,常温下继续反应。LC-MS监测反应完成后,将反应液倒入分液漏斗中,用EtOAc(3×50 mL)萃取、合并有机相,以饱和食盐水(5×100 mL)洗涤,无水硫酸钠干燥,过滤、减压旋干得到淡黄色黏稠物,以二氯甲烷复溶后,100~200目硅胶拌样,柱层析分离纯化(PE: EA=1:1)得中间体6 13.0 g,收率为70%。m.p.188~189℃,ESI-MSm/z: 931.27[M+H]+,C56H50O13,MW=930.33;1H NMR (600 MHz,DMSO-d6) δ 7.70(s,2H),7.61 (d,J=7.5 Hz,2H),7.54 (d,J=7.3Hz,4H),7.51 (d,J=7.3 Hz,2H),7.37(m,17H),6.92 (d,J=2.1 Hz,1H),6.75 (d,J=2.1 Hz,1H),5.49 (d,J=7.8 Hz,1H),5.35(s,1H),5.26~4.98 (m,6H),5.18 (d,J=11.8 Hz,2H),5.05 (q,J=11.2 Hz,2H),4.89 (s,1H),4.64 (s,1H),4.54(s,1H),3.71 (s,1H),3.67 (t,J=8.6 Hz,1H),3.57 (dd,J=14.0,8.9 Hz,1H),3.45 (m,3H)。
中间体7:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-(3′,4′-O-异丙基)半乳糖基-4H-苯并吡喃酮
将11.4 g(12 mmol)中间体6、800 mgp-TsOH.H2O、14 g无水CaSO4及200 mL丙酮加入反应瓶中,搅拌24 h,TLC监测反应完毕后,过滤除去CaSO4,滤液中加入少量Et3N终止反应,加入200~300目硅胶拌样,柱层析分离纯化(PE: EA=2:1)得中间体7 9.6 g,收率为83%。m.p.196℃~197℃,ESI-MSm/z: 971.46[M+H]+,C59H54O13,MW=970.36;1H NMR (600 MHz,DMSO-d6) δ 7.70 (s,2H),7.61 (d,J=7.5 Hz,2H),7.51(d,J=7.3 Hz,6H),7.35(m,17H),6.92 (d,J=2.1 Hz,1H),6.76 (d,J=2.1 Hz,1H),5.64 (d,J=4.5 Hz,1H),5.52 (d,J=8.2 Hz,1H),5.25~5.22 (m,6H),5.14 (d,J=11.7 Hz,2H),5.05 (q,J=11.2 Hz,2H),4.78 (t,J=5.5 Hz,1H),4.14 (dd,J=5.5,1.7 Hz,1H),4.10 (m,1H),3.84 (m,1H),3.63 (t,J=11.2,5.6 Hz,1H),3.57 (m,1H),3.50 (t,J=7.6 Hz,1H),1.20 (d,6H) 。
中间体8a-1:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′,6′′-2-O-(3-氟肉桂酰基)-3′′,4′-O-异丙基半乳糖基]-4H-苯并吡喃酮
将729 mg(0.75 mmol)化合物7、500 mg(3 mmol)间氟肉桂酸、1.2 g DCC(6 mmol)、360 mg(3 mmol)DMAP及20 mLCH2Cl2加入反应瓶中,室温搅拌过夜,LC-MS监测反应完成后,将反应液放置在-40℃低温浴槽中,过滤除去固体后,加入0.5 mol/L的柠檬酸溶液(3×20 mL)洗涤,有固体析出,过滤除去,用0.5 mol/L的NaHCO3(3×20 mL)洗涤,水洗至中性后,放置-40℃低温浴槽中,有固体析出,过滤除去,向滤液中加入100~200目硅胶拌样,柱层析分离纯化(CH2Cl2:MeOH=200:1)得粗产品,浓缩后用二氯甲烷溶解,用100~200目硅胶拌样,柱层析分离纯化(PE:EA=3:1)得533 mg中间体8a-1,收率为56%。m.p.137℃~138℃,ESI-MSm/z: 1267.84[M+H]+,C77H64F2O15,MW=1266.45;1H NMR (400 MHz,DMSO-d6) δ 7.76~7.64 (m,3H),7.60 (d,J=16.1 Hz,1H),7.61~7.42 (m,5H),7.35~7.02 (m,31H),6.76 (d,J=16.0 Hz,1H),6.63 (d,J=7.9 Hz,2H),6.49 (d,J=16.0 Hz,1H),6.64~6.52 (m,3H),5.85 (d,J=8.4 Hz,1H),5.11~4.98(m,11H),4.59~4.50 (m,1H),4.50 (s,1H),4.36 (d,J=9.0 Hz,1H),4.29 (d,J=6.8 Hz,2H),1.24 (s,6H)。
用此类方法可获得中间体8a-2~8a-8。
化合物8a-2:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′-2-O-(4-甲基肉桂酰基)-3′,4′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为55%,m.p.170℃~171℃,ESI-MSm/z:1259.85 [M+H]+,C79H70O15,MW=1258.47;1H NMR(400 MHz,DMSO-d6) δ 7.74 (d,J=15.9 Hz,1H),7.40~7.20 (m,36H),7.09 (d,J=7.8 Hz,2H),6.67 (d,J=5.9 Hz,2H),6.61 (d,J=16.0 Hz,1H),6.31 (d,J=16.0 Hz,1H),5.85 (d,J=8.4 Hz,1H),5.11~4.89 (m,11H),4.49~4.34 (m,2H),4.31~4.20 (m,3H),2.31 (s,3H),2.21 (s,3H),1.23 (s,6H)。
化合物8a-3:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′-2-O-(4-羟基基肉桂酰基)-3′′,4′′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为64%,m.p.131℃~132℃,ESI-MSm/z:1443.35 [M+H]+,C91H78O17,MW=1442.52;1H NMR(400 MHz,DMSO-d6) δ 7.74 (d,J=15.8 Hz,1H),7.45~7.08 (m,42H),7.01 (d,J=8.7 Hz,2H),6.95 (d,J=8.5 Hz,2H),6.69 (s,1H),6.63 (s,1H),6.51 (d,J=15.9 Hz,1H),6.24 (d,J=15.9 Hz,1H),5.85 (d,J=8.3 Hz,1H),5.11~4.86 (m,15H),4.52~4.34 (m,2H),4.34 (s,1H),4.27 (d,J=11.5 Hz,2H),1.23 (s,6H)。
化合物8a-4:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′′-2-O-(3,4,5-三甲氧基肉桂酰基)-3′,4′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为56%,m.p.100℃~101℃,ESI-MSm/z:1411.71 [M+H]+,C83H78O21,MW=1410.51;1H NMR(400 MHz,DMSO-d6) δ 7.66 (d,J=15.8 Hz,1H),7.43~7.11 (m,28H),7.03 (s,2H),6.82 (s,2H),6.69(d,J=15.9 Hz,1H),6.64 (s,1H),6.62 (s,1H),6.47 (d,J=15.9 Hz,1H),5.87 (d,J=8.3 Hz,1H),5.13~4.89 (m,11H),4.62~4.49 (m,1H),4.51~4.46 (m,1H),4.37 (s,1H),4.28 (d,J=11.4 Hz,2H),3.78 (s,6H),3.70 (s,6H),3.68 (s,3H),3.65 (d,J=8.3 Hz,3H),1.23 (s,6H)。
化合物8a-5:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′′-2-O-(3,4,5-三羟基苯甲酰基)-3′,4′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为31%,m.p.78℃~80℃,ESI-MSm/z:1815.67 [M+H]+,C115H98O21,MW=1814.66;1H NMR(400 MHz,DMSO-d6) δ 7.60 (s,2H),7.33~7.12 (m,57H),7.11 (s,2H),6.61 (s,1H),6.48 (s,1H),5.96 (d,J=8.6 Hz,1H),5.32 (t,J=8.0 Hz,1H),5.08~4.86 (m,17H),4.77~4.68 (m,5H),4.67~4.62 (m,1H),4.60 (s,1H),4.49 (d,J=8.5 Hz,2H),4.40 (s,1H),1.28 (s,6H)。
化合物8a-6:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′′-2-O-(4-三氟甲基肉桂酰基)-3′,4′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为57%,m.p.101℃~102℃,ESI-MSm/z:1367.33 [M+H]+,C79H64F6O15,MW=1366.41;1H NMR(400 MHz,DMSO-d6) δ 7.65~7.42 (m,8H),7.41~7.11(m,25H),6.94 (d,J=8.7 Hz,2H),6.86 (d,J=8.7 Hz,2H),6.65 (d,J=6.8 Hz,2H),6.51 (d,J=15.9 Hz,1H),6.24 (d,J=15.9 Hz,1H),5.85 (d,J=8.5 Hz,1H),5.11~4.89 (m,11H),4.54~4.48 (m,2H),4.29~4,18 (m,3H),1.23 (s,6H)。
化合物8a-7:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′-2-O-(4-氯肉桂酰基)-3′′,4′′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为46%,m.p.101℃~102℃,ESI-MSm/z:1301.29 [M+H]+,C77H64Cl2O15,MW=1300.24;1H NMR(400MHz,DMSO-d6) δ 7.46~7.24 (m,37H),6.69 (d,J=16.1 Hz,1H),6.65 (m,2H),6.42 (d,J=15.9 Hz,1H),5.85 (d,J=8.4 Hz,1H),5.11~4.86 (m,11H),4.59~4.55(m,1H),4.52 (s,1H),4.36 (s,1H),4.29 (d,J=10.5 Hz,2H),1.23 (s,6H)。
化合物8a-8:5,7-双(苄氧基)-2-(3,4,5-三(苄氧基)苯基)-3-β-D-[2′′,6′-2-O-(4-溴肉桂酰基)-3′′,4′′-O-异丙基半乳糖基]-4H-苯并吡喃酮
收率为57%,m.p.98℃~99℃,ESI-MSm/z:1387.85 [M+H]+,C77H64Br2O15,MW=1386.26;1H NMR(400 MHz,DMSO-d6) δ 7.46~7.19 (m,37H),6.69 (d,J=16.1 Hz,1H),6.65~6.58 (m,2H),6.42 (d,J=15.9 Hz,1H),5.85 (d,J=8.4 Hz,1H),5.11~4.88 (m,11H),4.59~4.54 (m,1H),4.52 (s,1H),4.36 (s,1H),4.29 (d,J=10.5 Hz,2H),1.23 (s,6H)。
目标化合物9a-1:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(3-氟苯并酰基))]半乳糖基-4H-苯并吡喃酮
将380 mg (0.3 mmol) 化合物8a-1、4 mL THF、2 mL CH3OH加入反应瓶中,加入200 μL 4 mol/L盐酸-二氧六环溶液,室温搅拌,TLC检测反应至完全,加入几滴三乙胺终止反应后,过滤,滤液加入200~300目硅胶拌样,柱层析分离纯化(CH2Cl2:CH3OH=100:1)得186 mg固体产物,收率为51%;取123 mg(0.1 mmol)所得化合物、20 mg 质量分数为10%的钯碳、5 mL THF及2.5 mL CH3OH投入反应瓶中,室温下常压催化氢化过夜,LC-MS监测反应至完全,采用0.25 μm有机膜过滤除去Pd-C,少量甲醇洗涤后,回收溶剂采用半制备高效液相(甲醇:水=75%:25%)纯化得65 mg目标化合物9a-1,收率为73%。m.p.70~72℃,ESI-MSm/z: 781.19[M+H]+,C39H34F2O15,MW=780.19;1H NMR (400 MHz,CD3OD)δ 7.25 (s,2H),7.09 (ddJ=14.6,7.3 Hz,2H),6.96 (dd,J=7.7 Hz,2H),6.81~6.70 (m,4H),6.22 (s,1H),6.06(s,1H),5.42 (d,J=8.2 Hz,1H),5.30 (t,J=9.0 Hz,1H),4.25~4.22 (m,1H),4.08 (dd,J=11.5,4.2 Hz,1H),3.82(d,J=3.4 Hz,1H),3.75 (ddd,J=11.5,9.2,3.9 Hz,2H),2.92 (t,J=16.9 Hz,3H),2.78 (dt,J=15.3,7.6 Hz,1H),2.73~2.67 (m,1H),2.61 (td,J=7.5,3.1 Hz,2H),2.37 (t,J=7.8 Hz,2H);13C NMR (126 MHz,CD3OD) δ 179.4,174.8,174.4,166.2,165.6,163.7,163.4,158.7,158.5,146.8,145.3,145.1,138.5,135.8,131.6,131.5,131.4,125.8,125.6,122.3,116.7,116.5,116.3,114.4,114.2,110.4,106.2,102.3,100.2,95.0,75.0,74.4,73.2,71.0,64.9,37.0,36.9,32.0,31.8。
用此类方法可获得目标化合物9a-2~9a-5。
化合物9a-2:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(4-甲基苯并酰基))]半乳糖基-4H-苯并吡喃酮
收率为71%,m.p.70℃~72℃,ESI-MSm/z: 773.46[M+H]+,C41H40O15,MW=772.24;1H NMR (400 MHz,CD3OD) δ 7.26 (s,2H),7.05 (d,J=7.6 Hz,2H),6.95 (d,J=7.6 Hz,2H),6.88~6.86 (m,4H),6.25 (d,J=1.7 Hz,1H),6.13 (s,1H),5.43 (d,J=8.0 Hz,1H),5.31 (dd,J=9.6,8.3 Hz,1H),4.21 (dd,J=11.5,7.9 Hz,1H),4.10(dd,J=11.5,4.3 Hz,1H),3.81 (d,J=3.4 Hz,1H),3.75(dd,J=9.8,3.4 Hz,1H),3.66 (dd,J=7.9,4.3 Hz,1H),2.90 (dd,J=12.8,7.1 Hz,2H),2.75~2.73 (m,2H),2.61(t,2H),2.38 (t,2H),2.24 (s,3H),2.11 (s,3H);13C NMR (126 MHz,CD3OD) δ 177.8,173.8,173.5,166.5,158.2,158.1,157.9,151.0,150.8,150.7,150.5,150.4,150.3,148.2,140.2,136.8,136.6,136.4,136.3,132.6,132.2,130.7,130.6,125.7,125.6,124.5,124.4,124.3,124.2,117.0,111.0,75.2,74.9,73.6,70.7,37.9,37.2,31.3,31.2,21.4,21.3。
化合物9a-3:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(4-羟基苯并酰基))]半乳糖基-4H-苯并吡喃酮
收率为78%,m.p.84℃~85℃,ESI-MSm/z: 777.20[M+H]+,C39H36O17,MW=776.20;1H NMR (400 MHz,CD3OD) δ 7.27 (s,2H),7.02 (d,J=8.4 Hz,2H),6.84 (d,J=8.5 Hz,2H),6.61 (dd,J=8.5,6.6 Hz,4H),6.29 (d,J=2.1 Hz,1H),6.15 (d,J=2.1 Hz,1H),5.44 (d,J=8.1 Hz,1H),5.32 (dd,J=9.8,8.0 Hz,1H),4.22 (dd,J=11.5,7.9 Hz,1H),4.08 (dd,J=11.5,4.4 Hz,1H),3.82~3.78 (m,1H),3.75 (dd,J=9.8,3.4 Hz,2H),3.68~3.66 (m,1H),2.86(t,3H),2.71~2.69 (m,2H),2.57 (td,J=7.5,2.6 Hz,2H),2.36 (s,2H);13C NMR (126 MHz,CD3OD) δ 177.6,174.1,173.9,167.1,164.2,159.3,158.7,151.3,150.9,148.7,140.7,139.7,139.3,137.1,137.0,136.9,136.8,130.7,130.7,129.9,125.1,125.0,124.8,124.6,122.7,111.5,106.5,103.2,100.9,95.7,75.6,75.4,74.1,71.2,65.5,32.1,32.0,22.2,22.1。
化合物9a-4:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′,6′-2-O-(3‴-(3,4,5-三甲氧基苯并酰基))]半乳糖基-4H-苯并吡喃酮
收率为78%,m.p.72℃~73℃,ESI-MSm/z: 925.64[M+H]+,C45H48O21,MW=924.27;1H NMR (400 MHz,CD3OD) δ 7.23 (s,2H),6.49 (s,2H),6.30 (s,2H),6.18 (d,J=1.8 Hz,4H),6.10 (d,J=1.7 Hz,1H),5.49 (d,J=8.1 Hz,1H),5.34 (m,1H),4.29~4.26 (m,1H),4.10 (dd,J=11.5,3.9 Hz,1H),3.85 (d,J=3.5 Hz,1H),3.79~3.76 (m,2H),3.74 (s,6H),3.72 (s,3H),3.69 (s,6H),3.54 (s,3H),2.94~2.92 (m,2H),2.78~2.76 (m,2H),2.56~2.53 (m,2H),2.39~2.36 (m,2H);13C NMR (126 MHz,CD3OD)δ 177.8,173.5,173.4,166.5,163.5,158.7,158.1,154.6,150.8,150.6,150.4,148.2,140.2,138.0,137.9,137.7,136.5,136.3,136.1,132.2,130.0,124.5,124.4,124.2,124.1,122.1,110.9,106.8,106.0,102.9,100.3,95.1,75.2,74.8,73.6,70.7,65.2,61.3,61.2,37.5,37.0,32.3,31.2,21.4,21.1。
化合物9a-5:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′,6′-2-O-(3,,4,5-三羟基苯甲酰基))]半乳糖基-4H-苯并吡喃酮
收率为77%,m.p.78℃~79℃,ESI-MSm/z: 785.51[M+H]+,C35H28O21,MW=784.11;1H NMR (400 MHz,CD3OD) δ 7.25 (s,2H),7.15 (s,2H),6.95 (s,2H),6.29(d,J=2.1 Hz,4H),5.78 (d,J=8.0 Hz,1H),5.47 (dd,J=9.8,8.1 Hz,1H),4.42 (dd,J=11.1,6.8 Hz,1H),4.33(dd,J=11.1,6.3 Hz,1H),4.01 (d,J=3.1 Hz,1H),3.95 (t,J=6.6 Hz,1H),3.90 (dd,J=11.1,6.8 Hz,1H);13C NMR(126 MHz,CD3OD) δ 177.2,166.9,164.0,159.1,158.6,150.7,148.8,148.7,148.6,142.4,142.2,140.7,137.6 137.4,137.3,137.2,137.1,136.9,136.7,135.8,125.1,124.9,124.7,122.9,122.7,121.9,112.1,102.4,100.8,95.5,75.7,75.4,74.4,70.8,63.9。
目标化合物10a-1:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(3-氟肉桂酰基))]半乳糖基-4H-苯并吡喃酮
将410 mg (0.3 mmol)化合物8a-6、4 mL四氢呋喃、2 mL甲醇加入反应瓶中,加入200 μL 4 mol/L盐酸-二氧六环溶液,室温搅拌,TLC检测反应至完全,加入几滴三乙胺终止反应后,过滤,滤液加入200~300目硅胶拌样,柱层析分离纯化(CH2Cl2:CH3OH=100:1)得131 mg固体产物,收率为33%;取63 mg (0.05 mmol)所得化合物及5 mL二氯甲烷投入反应瓶中,-78℃下用注射器滴加2 mL 1 mol/L BBr3的二氯甲烷溶液,反应液中有固体析出,LC-MS监测反应完全后,加入3 mL甲醇终止反应,将反应液倒入冰水中,过滤得淡黄色固体,采用半制备高效液相纯化(甲醇:水=75%:25%)得17 mg化合物10a-1,收率为46%。m.p.146℃~147℃,ESI-MSm/z: 877.57[M+H]+,C41H30F6O15,MW=876.15;1H NMR (400 MHz,CD3OD)δ 7.80~7.68 (m,3H),7.68~7.46 (m,4H),7.58~7.46(m,2H),7.47 (d,J=16.0 Hz,1H),7.22 (s,2H),6.77(d,J=16.1 Hz,1H),6.39 (d,J=16.0 Hz,1H),6.15 (s,1H),6.04 (s,1H),5.48~5.36 (m,2H),4.53 (dd,J=11.4,8.5 Hz,1H),4.24~4.20 (m,1H),3.94 (s,1H),3.88~3.80(m,2H);13C NMR (126 MHz,CD3OD) δ 177.8,166.6,166.5,165.9,163.1,158.2,157.7,149.9,147.8,143.2,139.8,139.1,138.8,136.1,135.9,135.7,135.4,133.4,131.8,129.6,129.2,129.1,126.3,126.0,124.1,124.0,123.8,123.6,122.5,121.6,110.5,105.5,99.9,94.6,75.1,73.4,70.5,65.9,64.9,31.2,31.1。用此类方法可获得目标化合物10a-2~10a-3。
化合物10a-2:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(3-氯肉桂酰基))]半乳糖基-4H-苯并吡喃酮
收率为49%,m.p.177℃~178℃,ESI-MSm/z:831.09 [M+Na]+,C39H30Cl2O15,MW=808.10;1H NMR(400 MHz,CD3OD) δ 7.73 (d,J=16.0 Hz,1H),7.59 (d,J=8.5 Hz,2H),7.39~7.26 (m,7H),7.22 (s,2H),6.64(d,J=16.0 Hz,1H),6.26 (d,J=16.0 Hz,1H),6.16 (d,J=1.9 Hz,1H),6.05 (d,J=1.9 Hz,1H),5.52 (d,J=8.0 Hz,1H),5.44~5.42 (m,1H),4.50 (dd,J=11.5,8.4 Hz,1H),4.23 (dd,J=11.6,4.0 Hz,1H),3.94 (d,J=3.2 Hz,1H),3.86~3.82 (m,2H);13C NMR (126 MHz,CD3OD)δ 178.7,166.9,166.7,165.9 163.1,158.1,157.7,149.9,149.8,147.8,143.8,143.7,139.8,136.1,135.9,135.7,135.3,134.1,133.9,130.3,130.2,129.7,129.6,124.2,124.1,123.9,123.7,121.6,120.4,119.6 110.5,105.5,99.9,94.6,75.1,74.9,73.4,70.5,64.8。
化合物10a-3:2-(3,4,5-三羟基苯基)-5,7-二羟基-3-β-D-[2′′,6′′-2-O-(3‴-(3-溴肉桂酰基))]半乳糖基-4H-苯并吡喃酮
收率为72%,m.p.172℃~173℃,ESI-MSm/z:919.99 [M+Na]+,C39H30Br2O15,MW=896.00;1H NMR(400 MHz,CD3OD) δ 7.73 (d,J=16.0 Hz,1H),7.60 (d,J=8.5 Hz,2H),7.39~7.26 (m,7H),7.22 (s,2H),6.64(d,J=16.0 Hz,1H),6.28 (d,J=16.0 Hz,1H),6.16 (d,J=1.9 Hz,1H),6.05 (d,J=1.9 Hz,1H),5.52 (d,J=8.0 Hz,1H),5.44~5.40 (m,1H),4.50 (dd,J=11.5,8.4 Hz,1H),4.23 (dd,J=11.6,4.0 Hz,1H),3.94 (d,J=3.1 Hz,1H),3.86~3.76 (m,2H);13C NMR (126 MHz,CD3OD)δ 177.7,166.8,166.4,164.2,161.5,156.6,149.9,149.8,145.1,143.9,143.4,136.7,135.8,135.5,135.2,134.1,133.8,133.6,133.2,131.8,131.7,130.3,130.2,129.6,129.4,124.1,120.4,118.5,117.7,108.5,104.3,100.9,98.4,93.2,73.5,72.9,71.7,69.4,63.1。
2.2 抗菌活性测试
采用微量肉汤稀释法测定14个化合物最小抑菌浓度(MIC),详细参考美国国家临床实验室标准化委员会(Clinical and Laboratory Standards Institute,CLSI)抗菌药物敏感性试验操作规程[17]。用MH肉汤配置化合物2倍工作浓度的母液,母液中化合物的浓度分别为256、128、64、32、16、8、4、2、1、0.5、0.25和0.125 μg/mL。吸取100 μL的化合物母液加到灭菌的96孔聚苯乙烯板中的第1至第12孔内,然后各孔加入100 μL含有受试菌液的MH肉汤,每孔总液量为200 μL。孔内药物终浓度依次分别为128、64、32、16、8、4、2、1、0.5、0.25、0.125和0.06 μg/mL,菌液终浓度均为105CFU/mL。实验对照药物为左氧氟沙星,阳性对照组:孔内含有左氧氟沙星与菌液。菌对照组:孔内仅接种菌液,不含药物。细菌培养在37℃孵育18~20 h后观测结果,肉眼观察或酶标仪测定A600值,以在小孔内完全抑制细菌生长的最低药物浓度为其最低抑菌浓度(MIC)。实验中所用菌株为耐万古霉素肠球菌ATCC51299、万古霉素敏感肠球菌ATCC29212、耐甲氧西林金黄色葡萄球菌ATCC43330、甲氧西林敏感性金黄色葡萄球菌ATCC29213、耐甲氧西林表皮葡萄球菌、甲氧西林敏感性表皮葡萄球菌。
3 结果与讨论
3.1 化合物合成
在杨梅苷的苄基保护实验中,发现当K2CO3作为碱时,杨梅苷5-羟基无法发生苄基化反应。可能原因是5位羟基与4位羰基形成分子内氢键造成5位羟基难以被苄基化。通过优化发现,当选用Cs2CO3时,杨梅苷能够以较好的产率实现完全苄基化。在脱除杨梅苷3位鼠李糖时,通过优化在不同的醇溶液的反应条件发现,与常用的甲醇、乙醇相比,采用异丙醇作为溶剂时,反应更高效,反应时间短,产率高。
由于糖供体α-溴代-2,3,4,6-四乙酰基半乳糖苷反应活性高,但是不稳定容易降解,实验过程中我们采用现制现用的方法来降低糖基给体对糖苷化反应的影响。另外,由于糖基化的产物与3-羟基五苄基杨梅苷原料难于在TLC分离开来,为了更加高效准确地判断糖苷化反应终点,本反应采用LC-MS 跟踪反应。另外,由于糖苷化反应过程中,未反应完全的3-羟基五苄基杨梅素难于与糖苷化产物分离开来,我们将糖苷化反应产物直接碱性条件下脱乙酰基,暴露出糖环的羟基后再分离纯化,更加易于剔除糖苷化反应未反应完全的原料,同时减少了分离步骤,便于操作,易于纯化。
3.2 化合物抗菌活性
根据CLSI指导原则的微量稀释法,对新合成的14个化合物进行了抗菌活性测试,测试结果如表2所示。通过对抗菌活性MIC数据可以看出,14个化合物对于粪肠球菌的MIC大于128 μg/mL,无论是敏感还是耐药菌株都无抗菌活性。化合物9a-1~9a-4和10a-1~10a-3对金黄色葡萄球菌和表皮葡萄球菌显示出较好的抗菌活性,MIC值范围为32~1 μg/mL。其中半乳糖苷类杨梅素衍生物9a-1对耐甲氧西林葡萄球菌ATCC43300的最小抑菌浓度达到1 μg/mL。当用苯甲酸酰基修饰半乳糖3,4-位羟基时,化合物9a-5的抗菌活性完全消失。新型糖苷类杨梅素衍生物对耐药金黄色葡萄球菌的抗菌活性要稍强于敏感菌株,该现象在10a-1~10a-3中较为明显,但是10a-1~10a-3抗菌活性整体都弱于衍生物9a-1~9a-4。另外,3位羟基酰化衍生物中,仅有11a-1显示出一定的抗菌活性,其他化合物抗菌活性无显著改善。化合物11a-1~11a-6对这金黄色葡萄球菌和表皮葡萄球菌的抗菌活性弱于上述糖苷类衍生物,进一步提示半乳糖苷元修饰3位羟基的重要性。通过以上研究结果证明,3位羟基是个重要活性修饰位点,可能对衍生物的抗菌活性起着重要作用。

表2 衍生物9a-1~9a-5、10a-1~10a-3、11a-1~11a-6和对照药物对标准菌株的MIC值Tab.2 The MIC values of derivatives 9a-1~9a-5,10a-1~10a-3,11a-1~11a-6 and control drug against bacterial strains
4 结论
本研究以杨梅苷作为原料设计了新的合成路线,并对合成中的关键反应条件进行了优化,构建了一条高效快捷的杨梅素衍生物的制备方法。以天然杨梅素作为母核,合成了14个杨梅素类衍生物,利用核磁和ESI-MS对其结构进行了表征。通过微量肉汤稀释法,对化合物进行了抗菌活性测试,发现8个抗菌活性显著提高的活性化合物。化合物9a-1~9a-4的对革兰阳性菌抗菌菌活性较好,其中9a-1半乳糖苷类杨梅素衍生物对耐甲氧西林葡萄球菌ATCC43300的最小抑菌浓度达到1 μg/mL。化合物的初步活性研究结果显示,黄酮醇3位羟基半乳糖修饰可以显著提高杨梅素的抗菌活性,为进一步开展具有抗菌作用的杨梅素衍生物的研究提供了新的线索。
猜你喜欢
华人时刊(2022年13期)2022-10-27
文苑(2019年20期)2019-11-16
西江月(2018年5期)2018-06-08
作文周刊·小学二年级版(2018年9期)2018-04-18
中学课程辅导·教学研究(2017年29期)2018-02-26
中国医药科学(2016年9期)2016-07-25
数理化学习·高一二版(2009年1期)2009-03-19
中国科技术语(2004年4期)2004-03-18
