
新型氮杂环卡宾催化剂的合成及其在烯烃的自由基氟烷基酰化反应中的应用研究
2022-10-11 14:03:16代海渝戴青松何美浩李青竹杨四琳田远航王亚鹏刘万聪李俊龙王启卫
合成化学 2022年9期
代海渝, 戴青松, 何美浩, 李青竹,, 杨四琳, 田远航, 王亚鹏, 刘万聪, 张 翔*, 李俊龙,*, 王启卫,4*
(1. 成都大学 机械工程学院,四川 成都 610106; 2. 成都大学 药学院(川抗所)抗生素研究与再评价四川省重点实验室,四川 成都 610052; 3. 中国科学院 成都有机化学研究所,四川 成都 610041; 4. 西华大学 理学院,四川 成都 610039)
氮杂环卡宾(NHC)作为一种重要的有机小分子催化剂,其在有机合成领域发挥着至关重要的作用[1-3]。近年来,NHC的应用研究主要围绕于极性化学中的极性翻转及极性保持反应[4-6]。随着研究的不断深入,研究人员相继报道了氮杂环卡宾通过自由基历程催化的多种化学反应,很好地突破了底物电性效应的制约,实现了多种更富挑战的反应化学[7-11]。在这些由自由基介导的反应过程中,衍生自噻唑骨架的氮杂环卡宾以其独特的反应活性受到了人们的广泛关注[12-17]。因此,围绕噻唑这一优势骨架开展催化剂的结构创新[18-19]对进一步开发NHC在自由基催化历程中的应用潜力具有重要的科学意义和实际价值。
然而,目前研究所使用的噻唑盐类卡宾前体结构还比较单一,尤其是氮原子上取代基的选取主要围绕芳基及链状的烷基而进行,因此仍拥有较大的设计改造空间(Scheme 1a)。基于本课题组前期对氮杂环卡宾-金属催化剂改造的工作基础[20-21],本文采用环庚酮及环己(庚)胺为起始原料,经溴代、环化和脱硫等反应过程,以良好的收率合成了两种类型环烷基取代的新型噻唑骨架的氮杂环卡宾催化剂(Scheme 1b)。此外,本课题组发展的烯烃自由基氟烷基酰化反应为模板反应[22],对其催化活性进行了初步的评价,以中等的收率完成了3种类型的γ-氟烷基取代酮的高效合成(Scheme 1c)。所得化合物通过1H NMR,13C NMR,19F NMR和HR-MS(ESI-TOF)进行表征。

Scheme 1
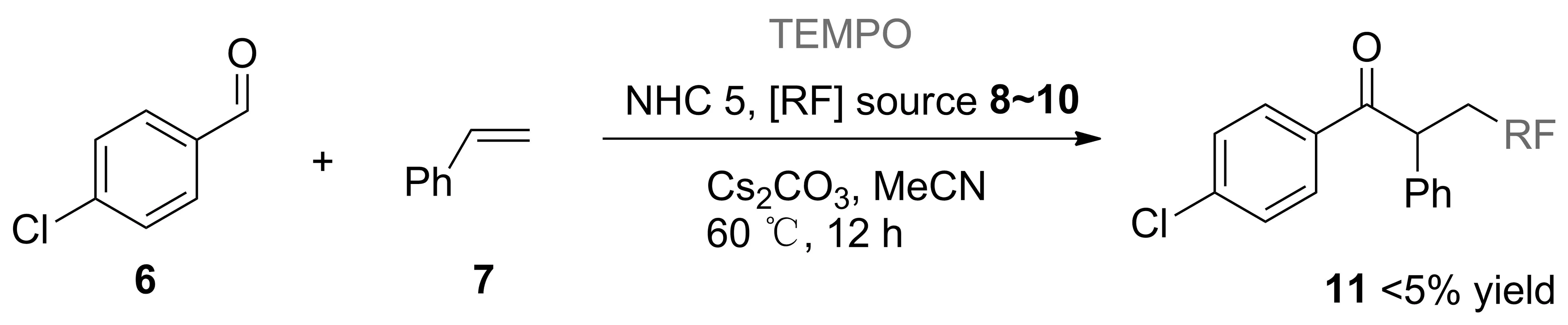
Scheme 2

Scheme 3
1 实验部分
1.1 仪器与试剂
WRX-X-4A型熔点仪;JEOL-600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters SYNAPT G2型高分辨质谱仪。
所用试剂均为分析纯。
1.2 NHC催化剂前体(5)的合成
(1)α-溴代环庚酮(2)的合成
向100.00 mL圆底烧瓶中加入环庚酮(48.00 mmol, 5.38 g),再加入60.00 mL二氯甲烷溶解。0 ℃下依次加入一水合对甲苯磺酸(4.80 mmol, 913.00 mg)和N-溴代丁二酰亚胺(48.00 mmol, 8.54 g)。于室温反应12 h(TLC监测环庚酮消耗完毕),在反应液中加入水,淬灭反应后使用二氯甲烷萃取,收集有机相,无水硫酸钠干燥,浓缩。经硅胶柱层析(洗脱剂:V石油醚/V乙酸乙酯=200/1)纯化得7.80 g产物2,淡黄色液体,收率85%;1H NMR(CDCl3, 600 MHz)δ: 4.32(dd,J=9.6 Hz, 5.4 Hz, 1H), 2.83~2.72(m, 1H), 2.50~2.38(m, 1H), 2.36~2.26(m, 1H), 1.99~2.26(m, 3H), 1.75~1.66(m, 1H), 1.56~1.45(m, 2H), 1.36~1.28(m, 1H);13C NMR(CDCl3, 150 MHz)δ: 206.0, 53.6, 39.2, 34.1, 29.4, 26.6, 24.8; HR-MS(ESI-TOF)m/z: calculated for C7H1179BrO{[M+Na]+}212.9885, found 212.9888; C7H1181BrO{[M+Na]+}214.9865, found 214.9869。
(2) 3-环己基环庚烷并噻唑-2-硫酮(4a)的合成
向50 mL圆底烧瓶中加入环己胺(13.00 mmol, 1.29 g),再加入7.80 mL DMSO溶解,随后加入20 N氢氧化钠水溶液(13.00 mmol, 0.65 mL), 0 ℃下加入二硫化碳(13.00 mmol, 0.99 g),于室温反应1 h后,再降温至0 ℃下加入2(13.00 mmol, 2.48 g),随后,再于室温下反应12 h。反应结束后(TLC监测环己胺消耗完毕),0 ℃下在反应液中加入水,乙酸乙酯萃取,收集有机相,无水硫酸钠干燥,浓缩后加入6.50 mL乙醇,0.65 mL浓盐酸,回流1 h后析出固体,过滤得产物4a2.47 g。
3-环己基环庚烷并噻唑-2-硫酮(4a):白色固体,收率71%, m.p.141.7~145.4 ℃;1H NMR(CDCl3, 600 MHz)δ: 5.71~5.64(m, 1H), 2.98~2.89(m, 2H), 2.54~2.47(m, 2H), 1.94~1.72(m, 9H), 1.68~1.58(m, 4H), 1.56~1.44(m, 2H), 1.26~1.12(m, 1H);13C NMR(CDCl3, 150 MHz)δ: 184.7, 142.7, 124.1, 59.1, 31.5, 30.2, 28.9, 26.9, 26.5, 26.1, 25.8, 25.3; HR-MS(ESI-TOF)m/z: calculated for C14H21NS2{[M+H]+}268.1188,found 268.1193。
(3) 3-环庚基环庚烷并噻唑-2-硫酮(4b)的合成
50 mL圆底烧瓶中加入环庚胺(13.0 mmol, 1.47 g),再加入7.8 mL DMSO溶解,随后加入20 N氢氧化钠水溶液(13.00 mmol, 0.65 mL)。 0 ℃下加入二硫化碳(13.00 mmol, 0.99 g),于室温反应1 h后,再降温至0 ℃并加入α-溴代环庚酮2(13.00 mmol, 2.48 g),随后,再于室温下反应12 h。反应结束后(TLC监测环己胺消耗完毕),于0 ℃下向反应液中加入水,乙酸乙酯萃取,收集有机相,无水硫酸钠干燥,浓缩后加入6.50 mL乙醇,0.65 mL浓盐酸,回流1 h后析出固体,过滤得产物4b2.49 g。
3-环庚基环庚烷并噻唑-2-硫酮(4b):淡黄色固体,收率68%, m.p.109.2~111.7 ℃;1H NMR(CDCl3, 600 MHz)δ: 5.89~5.82(m, 1H), 2.85~2.79(m, 2H), 2.53~2.47(m, 2H), 1.97~1.80(m, 6H), 1.79~1.59(m, 10H), 1.58~1.49(m, 2H);13C NMR(CDCl3, 150 MHz)δ: 183.8, 142.2, 124.1, 60.2, 32.4, 31.5, 28.9, 28.0, 27.0, 26.9, 26.6, 25.9; HR-MS(ESI-TOF)m/z: calculated for C15H23NS2{[M+H]+}282.1345, found 282.1341。
(4) 氮杂环卡宾催化剂前体(5a)的合成
取3-环己基环庚烷并噻唑-2-硫酮4a(8.63 mmol, 2.31 g)加入100 mL圆底烧瓶中,加入35.00 mL冰乙酸溶解,0 ℃下逐滴加入30%双氧水(28.48 mmol, 3.23 g)搅拌1 h。蒸除溶剂,用6 mL甲醇溶解,于0 ℃下加入高氯酸钠甲醇水溶液(34.87 mmol, 4.27 g, 20.00 mL甲醇, 10.00 mL水),再将反应移至室温下反应1 h。反应结束后(TLC监测4a消耗完毕),在反应液中加入水,二氯甲烷萃取,收集有机相,无水硫酸钠干燥,浓缩后加入乙醚析出固体,过滤,得到1.85 g氮杂环卡宾催化剂前体5a。
氮杂环卡宾催化剂前体(5a):白色固体,收率64%, m.p.175.6~178.8 ℃;1H NMR(CDCl3, 600 MHz)δ: 9.70(s, 1H), 4.38~4.32(m, 1H), 3.02~2.97(m, 4H), 2.18(d,J=12.6 Hz, 2H), 2.04~1.94(m, 4H), 1.89~1.64(m, 8H), 1.54~1.45(m, 2H);13C NMR(CDCl3, 150 MHz)δ: 151.0, 147.8, 139.9, 63.8, 33.2, 30.6, 27.7, 26.9, 26.2, 25.3, 25.0, 24.5; HR-MS(ESI-TOF)m/z: calculated for C14H22NS{[M]+}236.1467, found 236.1469。
(5) 氮杂环卡宾催化剂前体(5b)的合成
取3-环庚基环庚烷并噻唑-2-硫酮4b(8.42 mmol, 2.37 g)加入100 mL圆底烧瓶中,使用35.00 mL冰乙酸溶解,在0 ℃下逐滴加入30%双氧水(27.77 mmol, 2.84 g)并搅拌1 h。随后蒸除溶剂,用6 mL甲醇溶解,于0 ℃下加入高氯酸钠甲醇水溶液(34.52 mmol, 4.23 g, 20 mL甲醇, 10 mL水),再将反应移至室温下反应1 h。反应结束后(TLC监测4b消耗完毕),在反应液中加入水,二氯甲烷萃取,收集有机相,无水硫酸钠干燥,浓缩后加入乙醚析出固体,过滤,得到1.80 g氮杂环卡宾催化剂前体5b。
氮杂环卡宾催化剂前体(5b):淡黄色固体,收率61%, m.p.169.5~172.3 ℃;1H NMR(CDCl3, 600 MHz)δ: 9.71(s, 1H), 4.57~4.45(m, 1H), 3.02~2.97(m, 4H), 2.22~2.15(m, 2H), 2.10~2.02(m, 2H), 2.00~1.94(m, 2H), 1.93~1.87(m, 2H), 1.86~1.80(m, 4H), 1.72~1.65(m, 4H), 1.64~1.56(m, 2H);13C NMR(CDCl3, 150 MHz)δ: 151.2, 147.6, 139.8, 66.1, 35.3, 30.5, 27.7, 27.0, 26.7, 26.2, 25.0, 24.3; HR-MS(ESI-TOF)m/z: calculated for C15H24NS{[M]+}250.1624, found 250.1630。
1.3γ-氟烷基取代酮(11)的合成(以5a为例)
(1) 1-(4-氯苯基)-4,4,4-三氟-2-苯基丁烷-1-酮11a的合成
氩气条件下,10 mL封管中依次加入对氯苯甲醛(0.10 mmol, 14.10 mg)、苯乙烯(0.15 mmol, 15.60 mg)、 Togni CF3试剂(0.20 mmol, 66.00 mg)、催化剂5a(0.01 mmol, 3.40 mg)、碳酸铯(0.02 mmol, 6.50 mg)和1。00 mL乙腈,60 ℃下反应12 h。反应结束后(TLC监测产物对氯苯甲醛消耗完毕),蒸除溶剂,残余物经硅胶柱层析(洗脱剂:V石油醚/V乙酸乙酯=150/1)纯化得23.30 mg产物11a。
1-(4-氯苯基)-4,4,4-三氟-2-苯基丁烷-1-酮(11a):白色固体,收率74%, m.p.66.3~69.2 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.89(d,J=9.0 Hz, 2H), 7.38(d,J=8.4 Hz, 2H), 7.32(t,J=7.2 Hz, 2H), 7.29~7.23(m, 3H), 4.83(dd,J=7.2 Hz, 5.4 Hz, 1H), 3.33~3.25(m, 1H), 2.58~2.48(m, 1H);13C NMR(CDCl3, 150 MHz)δ: 195.5, 139.9, 137.1, 134.0, 130.2, 129.4, 129.0, 128.0, 128.0, 127.2, 47.3, 37.3(q,J=28.8 Hz, 1C);19F NMR(CDCl3, 564 MHz)δ: -65.03(t,J=11.3 Hz, 3F); HR-MS(ESI-TOF)m/z: calculated for C16H1235ClF3O{[M+H]+}313.0602, found 313.0599; C16H1237ClF3O{[M+H]+}315.0572, found 315.0575。
(2) 5-(4-氯苯基)-2,2-二氟-5-氧代-4-苯基戊酸乙酯(11b)的合成
氩气条件下,10.00 mL封管中依次加入对氯苯甲醛(0.10 mmol, 14.10 mg)、苯乙烯(0.15 mmol, 15.60 mg)、二氟溴乙酸乙酯(0.20 mmol, 40.60 mg)、催化剂5a(0.01 mmol, 3.40 mg)、碳酸铯(0.15 mmol, 48.90 mg)和1.00 mL乙腈,60 ℃下反应12 h。反应结束后(TLC监测产物对氯苯甲醛消耗完毕),蒸除溶剂,残余物经硅胶柱层析(洗脱剂:V石油醚/V乙酸乙酯=50/1)纯化得16.50 mg产物11b。5-(4-氯苯基)-2,2-二氟-5-氧代-4-苯基戊酸乙酯(11b):淡黄色液体,收率45%;1H NMR(CDCl3, 600 MHz)δ: 7.88(d,J=8.4 Hz, 2H), 7.36(d,J= 9.0 Hz, 2H),7.30(t,J=7.2 Hz, 2H), 7.26(d,J=7.8 Hz, 2H), 7.23(t,J=6.6 Hz, 1H), 4.96(dd,J=7.8 Hz, 4.8 Hz, 1H), 4.21~4.14(m, 1H), 4.11~4.04(m, 1H), 3.31~3.20(m, 1H), 2.57~2.47(m, 1H), 1.24(t,J= 7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 196.1, 163.7(t,J=31.7 Hz, 1C), 139.7, 137.5, 134.1, 130.2, 129.3, 129.0, 128.2, 127.8, 115.2(t,J=249.9 Hz, 1C), 63.0, 47.0, 38.1 (t,J=23.0 Hz, 1C), 13.8;19F NMR(CDCl3, 564 MHz)δ: -104.55(dt,J=259.4 Hz, 15.2 Hz, 1F), -105.17(dt,J=260.0 Hz, 17.5 Hz, 1F); HR-MS(ESI-TOF)m/z: calculated for C19H1735ClF2O3{[M+H]+}367.0907, found 367.0908; C19H1737ClF2O3{[M+H]+}369.0878, found 369.0883。
(3) 1-(4-氯苯基)-4,4,5,5,6,6,7,7,8,8,9,9,9-十三氟-2-苯基壬烷-1-酮(11c)的合成
氩气条件下,10.00 mL封管中依次加入对氯苯甲醛(0.10 mmol, 14.10 mg)、苯乙烯(0.15 mmol, 15.60 mg)、全氟己基碘烷(0.20 mmol, 89.20 mg)、催化剂5a(0.01 mmol, 3.40 mg)、碳酸铯(0.15 mmol, 48.90 mg)和1.00 mL乙腈,并于60 ℃下反应12 h。反应结束后(TLC监测产物对氯苯甲醛消耗完毕),蒸除溶剂,残余物经硅胶柱层析(洗脱剂:V石油醚/V乙酸乙酯=180/1)纯化得18.00 mg产物11c。
1-(4-氯苯基)-4,4,5,5,6,6,7,7,8,8,9,9,9-十三氟-2-苯基壬烷-1-酮(11c):白色固体,收率32%, m.p.89.1~93.3 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.91(d,J=9.0 Hz, 2H), 7.38(d,J=7.8 Hz, 2H), 7.35~7.28(m, 4H), 7.26(t,J=7.2 Hz, 1H), 4.96(dd,J=8.4 Hz, 4.2 Hz, 1H), 3.51~3.38(m, 1H), 2.49~2.38(m, 1H);13C NMR(CDCl3, 150 MHz)δ: 195.5, 139.9, 137.3, 133.9, 130.2, 129.5, 129.0, 128.0, 128.0, 122.5~108.2(m, 6C), 45.7, 34.3(t,J=31.7 Hz, 1C);19F NMR(CDCl3, 564 MHz)δ: -81.21(t,J=8.5 Hz, 3F), -112.08~-112.75(m, 1F), -113.28~-114.00(m, 1F), -122.21(brs, 2F), -123.28(brs, 2F), -123.90(brs, 2F), -126.47~-126.71(m, 2F); HR-MS(ESI-TOF)m/z: calculated for C21H1235ClF13O{[M+Na]+}585.0261, found 585.0253; C21H1237ClF13O{[M+Na]+}587.0232, found 587.0228。
2 结果与讨论
2.1 NHC催化剂(5)的催化性能
基于设计合成的两种环烷基取代的噻唑卡宾催化剂,以本课题组发展的烯烃自由基氟烷基酰化反应作为模板反应,对催化剂的催化活性进行了考察。据表1所示,当使用Togni试剂8作为三氟甲基源时,无论是环己基取代的NHC催化剂5a,还是环庚基取代的5b,均能够以良好的收率得到目标产物γ-三氟甲基取代酮11a(Entry 1~2)。然而进一步使用二氟溴乙酸乙酯9作为二氟烷基源(Entry 3~4)时目标化合物的收率降低。其原因可能为9的氧化性较弱,不能够充分氧化Breslow中间体。但本实验仍能以中等收率(45%~48%)得到γ-二氟烷基取代酮。在此基础上,反应活性相对较低的全氟己基碘烷也被作为多氟烷基源进行考察(Entry 5~6)。结果表明,NHC催化剂5也能催化该烯烃自由基氟烷基酰化反应。通过上述3种不同的烯烃自由基氟烷基酰化反应,初步论证了设计合成的NHC催化剂具有良好的催化活性。
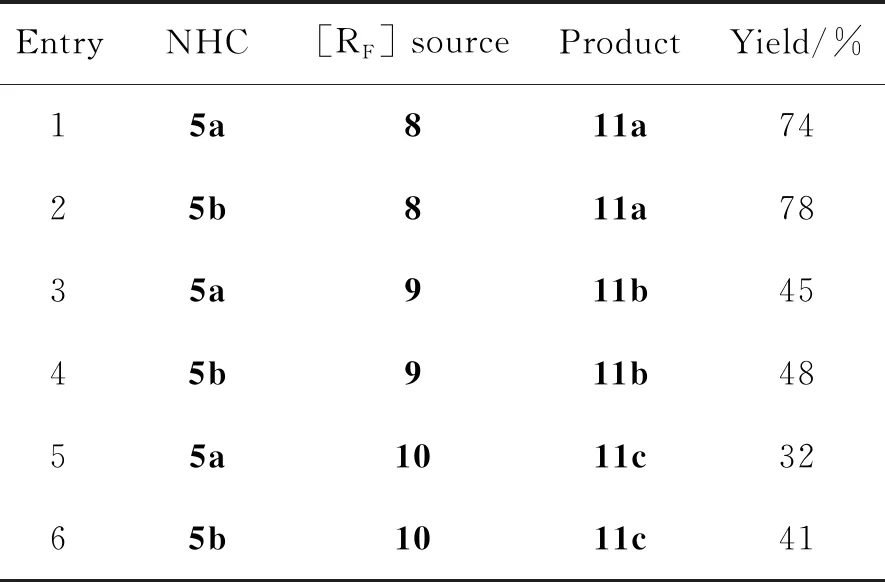
表1 NHC催化剂5的催化性能研究
2.2 烯烃的氟烷基酰化的反应机理
在上述活性评价的基础上,通过自由基抑制实验对反应的机理进行了初步的研究。结果发现,无论使用NHC催化剂5a或5b进行催化反应,当向反应体系中加入自由基抑制剂TEMPO时,反应均受到抑制,无法给出相应的目标产物(Scheme 2)。同时结合已有文献[22],可以认为上述催化过程为NHC介导的自由基催化历程。
据此,提出了上述烯烃氟烷基酰化反应可能的反应机理(Scheme 3):在碱的作用下,噻唑高氯酸盐5被活化产生氮杂环卡宾,与芳醛形成去质子Breslow中间体I;随后I与氟烷基试剂通过单电子还原过程产生氟烷基自由基A,对烯烃进行自由基加成,然后通过自由基偶联得到两性离子中间体II;最后,NHC离去的同时释放γ-氟烷基取代酮,完成催化循环。
以环庚酮及环己(庚)胺为起始原料,设计合成了两种环烷基取代的新型噻唑骨架的氮杂环卡宾催化剂。并通过烯烃的自由基氟烷基酰化反应,在完成了3种类型的γ-氟烷基取代酮的高效合成的同时,对其催化活性进行评价与论证。为通过自由基历程的氮杂环卡宾化学提供了一种新型的催化工具,有助于进一步开发NHC在自由基化学领域中的应用潜力,相关深度应用研究仍在进一步进行中。
猜你喜欢
云南化工(2021年7期)2021-12-21 07:27:22
科学技术与工程(2020年34期)2021-01-08 05:43:32
世界农药(2019年4期)2019-12-30 06:25:08
合成化学(2015年2期)2016-01-17 09:03:25
合成化学(2015年9期)2016-01-17 08:57:21
郑州大学学报(工学版)(2015年1期)2015-03-24 00:55:36
时尚北京(2015年1期)2015-01-30 00:00:35
无机化学学报(2014年6期)2014-02-28 17:31:59
无机化学学报(2014年1期)2014-02-28 17:30:01
中国兽药杂志(2012年4期)2012-11-06 07:26:18
