
三种DNA 提取方法对半滑舌鳎肠道菌群高通量测序结果的影响
2022-10-10 03:50:30葛仑华江博赟孙敬锋吕爱军胡秀彩陈丽梅
天津农学院学报 2022年3期
葛仑华,江博赟,孙敬锋,吕爱军,胡秀彩,陈丽梅
(天津农学院 水产学院,天津 300392)
半滑舌鳎(Cynoglossus semilaevis)具有肉质鲜美、个体大、生长快等优良性状,是我国重要的海水养殖鱼类[1]。近年来随着工厂化、集约化养殖模式的发展,半滑舌鳎病害发生频繁,严重降低了养殖的成活率[2]。肠道菌群在水产动物健康、生长、生理等多个方面发挥了重要作用,其结构组成与多样性对宿主健康具有重要影响[3-4]。对半滑舌鳎肠道菌群的研究有助于了解宿主与其肠道菌群之间的相互作用关系,并为疾病监控及预防提供新思路[5-7]。
近年来,越来越多的研究集中于分析水生动物肠道微生物群落结构[8]。早期研究主要是对获得的纯培养菌株进行鉴定分析,但这种方法的应用局限于可培养微生物,只能鉴定到少数的细菌群落,无法深入了解菌群组成结构[9]。随着现代生命科学技术的飞速发展,使用高通量测序技术可以同时对数百万个DNA 分子进行测序,通过分析微生物的单一基因或全基因组,可以鉴定出大部分肠道菌群的类别[10]。目前具有代表性的高通量测序技术主要有454 焦磷酸测序、Solexa 聚合酶合成测序、SOLiD 连接酶测序[11]。以Roche 454 和Illumina MiSeq 为代表的高通量测序平台也已应用在半滑舌鳎[5]、对虾(Penaeus monodon)[12]、尼罗罗非鱼(Oreochromis niloticus)[13]等多种水生动物肠道菌群结构的分析。
高通量测序技术的发展和应用为肠道菌群结构分析研究提供了技术支持。如何高质量地从样品中获得微生物基因组DNA是运用高通量测序技术分析肠道菌群的关键步骤。理想的微生物DNA提取方法既要考虑到DNA 产量和纯度,又要尽量避免DNA 的降解及降低试剂材料的成本[14]。目前有很多用于提取DNA 的试验方法,如酶促DNA提取技术[15]、超声波破碎技术[16]、玻璃珠机械裂解技术[17]、磁性分离技术[18]。
不同试验方法提取的DNA,经过测序鉴定后,对应的菌群结构分析结果存在偏差[19]。温崇庆等[20]用三种不同DNA 提取试剂盒(Omega,美国)提取凡纳滨对虾(Litopenaeus vannamei)的肠道菌群DNA,并经高通量测序分析,得到的肠道菌群组成结构分析结果差异显著。本试验室曾以溶菌酶结合超声波裂解法(Combination of Lysozyme and Ultrasonic Lysis,CLU)、Zirmil-beating 细胞破碎法(Zirmil-beating Cell Disruption,ZBC)和德国Qiagen 公司的QIA 试剂盒法(QIA)三种方法提取锦鲤(Cyprinus carpio)肠道菌群DNA,发现其高通量测序结果存在差异[21]。但目前不同DNA 提取方法对半滑舌鳎肠道菌群结构分析结果的影响未见报道。
本研究以CLU、ZBC 和QIA 三种方法提取半滑舌鳎肠道菌群DNA,通过高通量测序技术分析不同DNA提取方法引起的肠道菌群结构分析结果偏差,旨在为半滑舌鳎肠道菌群多样性研究方法,特别是DNA 提取策略提供参考资料,同时有助于全面了解健康半滑舌鳎肠道菌群结构特征。
1 材料与方法
1.1 半滑舌鳎肠道微生物收集
试验用半滑舌鳎购于天津市某养殖场,体长(30.0±2.5)cm,体重(220.0±6.5)g,将鱼运至天津农学院水产动物疾病及免疫学实验室,在温度为20 ℃,盐度为30 的海水中暂养一周,每日投喂颗粒饲料两次。选取3 尾健康半滑舌鳎用于肠道微生物的收集。首先用75%酒精棉球擦拭鱼体表,然后用无菌解剖刀剖开腹腔。从腹腔取出完整肠道,并将肠内容物收集至50 mL 离心管中。将样品与10 mL 磷酸盐缓冲溶液(PBS,0.01 mol/L,pH 7.2)混合,涡旋震荡30 s,然后将悬浊液在4 ℃,150 g 离心5 min,将上清液转移到新的无菌50 mL 离心管中。随后在4 ℃,2 700 g离心5 min,弃上清后获得沉淀。最后用3 mL PBS重新悬浮沉淀,将肠道菌群悬液平均分为三份,每份1 mL,分别用CLU、ZBC、QIA 三种方法进行DNA 提取。
1.2 DNA 的提取
1.2.1 CLU 方法
参照HAN等[22]应用CLU方法提取肠道菌群基因组DNA,将DNA 沉淀用1 mL 70%乙醇洗涤两次,风干,随后用100 μL 50 ℃预热的TE 缓冲液溶解。DNA 放置于-80 ℃保存备用。
1.2.2 ZBC 方法
参照JIANG 等[23]应用ZBC 方法提取菌群基因组DNA,最后将EP 管倒置15 min,待DNA 沉淀风干,然后用100 μL TE 缓冲液溶解DNA。DNA放置于-80 ℃保存备用。
1.2.3 QIA 方法
将1 mL 肠道菌群悬液在4 ℃,2 700 g 离心5分钟,用220 μL PBS 重悬细菌沉淀。然后应用QIAamp Fast DNA Stool Mini Kit(Qiagen,德国)提取肠道菌群DNA。最后用100 μL TE 缓冲液洗脱DNA。提取的DNA 放置于-80 ℃保存备用。
1.3 16S rDNA 高通量测序
选取16S rRNA 的V3-V4 可变区基因进行PCR 扩增,PCR 扩增所用的引物为341F(5′-CCC TACACGACGCTCTTCCGATCTGCCTACGGGNGGC WGCAG-3′)和805R(5′-GACTGGAGTTCCTTG GCACCCGAGAATTCCAGACTACHVGGGTATCTA ATCC-3′)。PCR 反应体系包含10×PCR buffer 5 μL,dNTP(10 mmol/L)0.5 μL,模板DNA 10 ng,正反向引物(50 μmol/L)各0.5 μL,Plantium Taq(5 U/μL)0.5 μL,加无菌超纯水至50 μL。PCR扩增反应条件如下:94 ℃ 3 min;94 ℃ 30 s,45 ℃ 20 s,65 ℃ 30 s,5 个循环;94 ℃ 20 s,55 ℃ 20 s,72 ℃ 30 s,20 个循环;72 ℃ 5 min。然后引入Illumina 桥式PCR 兼容引物进行第二轮扩增,反应体系包括10×PCR buffer 5 μL,dNTP(10 mmol/L)0.5 μL,模板DNA 20 ng,正反向引物(50 μmol/L)各0.5 μL,Plantium Taq(5 U/μL)0.5 μL,加无菌超纯水至50 μL。PCR 反应条件为:95 ℃ 30 s,95 ℃ 15 s,55 ℃ 15 s,72 ℃ 30 s,5 个循环;72 ℃ 5 min。PCR 产物经2%琼脂糖凝胶电泳检测,最后对PCR 产物进行纯化回收,使用Qubit 2.0 核酸和蛋白定量仪(Thermo Scientific,美国)对DNA 含量进行测定。在每个肠道菌群DNA 样品中掺入独特的条形码,利用Illumina MiSeq 平台进行测序。
1.4 数据处理分析
完成测序后,根据样品独特的条形码将测序读数分配给每个样品。数据分析的方法参照HAN等[21],首先使用FLASH 合并来自原始DNA 片段的成对读数[24],然后使用质量控制程序,包括剪切条形码和引物、通过PRINSEQ 软件过滤低质量读数、校正潜在的测序错误、应用UCHIME 去除嵌合体[25]。最后,计算Rarefaction,Chao1 和Simpson指数进行Alpha 多样性分析,计算加权UniFrac 度量距离进行Beta 多样性分析。为了得到每个OTU对应的物种分类信息,采用核糖体数据库项目(RDP)分类器[26]对97%相似度水平的OTU 代表序列进行分类学分析,并在门、属水平,统计各个样品的菌落组成。
2 结果与分析
2.1 质控与OTU 聚类分析
将CLU、ZBC、QIA 三种方法提取的DNA 样品用于高通量测序分析,得到的测序序列读数(filtered-number)分别为32 215、38 745、25 122(表1)。这些序列的平均长度分别为420.0 bp、417.0 bp、418.7 bp。根据97%的相似性聚类后,所有肠道细菌样品共发现7 516 个OTU(图1)。CLU、ZBC、QIA 方法对应样品中的OTU 数量分别为3 761、3 859、2 850(表1),其中三个样品共有的OTU数量为881,分别占三种样本各自OTU数量的23.42%、22.82%、30.91%。
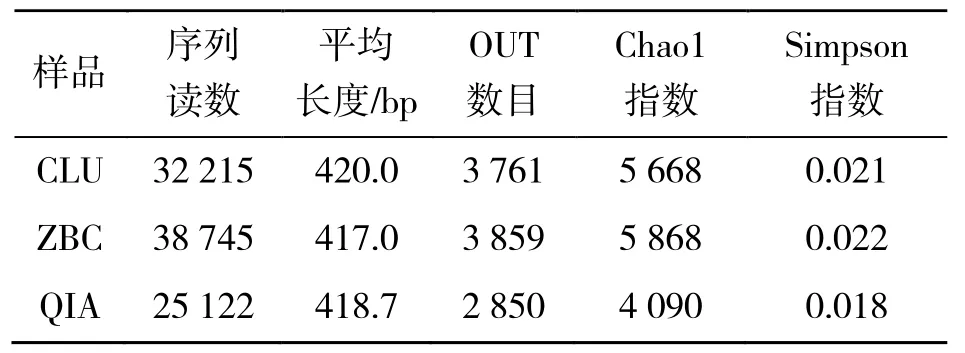
表1 三种方法提取的DNA样品的序列读数、OTU、Alpha多样性指数

图1 三种DNA 提取方法对应样品中的OTU 数目
2.2 Alpha 多样性分析
三种方法对应样本的稀释曲线都接近平缓,其中CLU 和ZBC 对应样本呈现相似的趋势,与QIA 相比,两者曲线斜率更大(图2a)。在Alpha多样性分析中,CLU、ZBC、QIA 方法对应的Chao1指数分别为5 668、5 868、4 090(图2b),Simpson指数分别为0.021、0.022、0.018(图2c),QIA 方法对应的Chao1 和Simpson 指数均低于CLU 和ZBC 方法。

图2 Alpha 多样性分析
2.3 半滑舌鳎肠道菌群结构组成分析
在门的水平上,三种方法提取的DNA 样品中共鉴定出31 个细菌门。其中CLU、ZBC、QIA 方法对应样品分别鉴定到26、29、28 个细菌门,共有的细菌门25 个。10 个丰度最高的优势菌门为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、螺旋菌门(Spirochaetae)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、疣微菌门(Verrucomicrobia)、芽单胞菌门(Gemmatimonadetes)(表2)。在CLU、ZBC、QIA 对应的样品中,这10 个优势菌门均被鉴定到,且大部分序列属于前四种细菌门(变形菌门、厚壁菌门、螺旋菌门、拟杆菌门),所占比例分别为85.94%、86.88%、83.45%(图3)。6 个存在差异的菌门为嗜热丝菌门(Caldiserica)、奇古菌门(Thaumarchaeota)、脱铁杆菌门(Deferribacteres)、软壁菌门(Tenericutes)、待定菌群(Candidate_division_OD1 和 Candidate_division_WS3)。这些类群均为低丰度菌群,这表明三种方法对一些丰度较低的菌群类别鉴定结果存在偏差。

图3 三种方法提取的DNA 样品中优势菌门相对丰度

表2 三种方法提取的DNA样品间优势菌门和OTU丰度比较
在属的水平上,所有样品序列共鉴定出799个属。CLU、ZBC、QIA 样品分别鉴定到593、599、495 个属,三个样品共有的细菌属362 个。以占样品平均丰度≥1%的属作为优势菌属,CLU 和ZBC各有20 个优势菌属,其中有14 个优势菌属为两者共有;而QIA 有19 个优势菌属,与CLU、ZBC均共享13 个优势菌属,三个样品之间共有的优势菌属为12 个。其中10 个最高丰度的菌属为短螺旋体属(Brevinema)、微小杆菌属(Exiguobacterium)、乳杆菌属(Lactobacillus)、伯克氏菌属(Burkholderia)、芽孢杆菌属(Bacillus),假单胞菌属(Pseudomonas)、肉杆菌属(Carnobacterium)、类似芽球菌属(Blastocatella)、乳球菌属(Lactococcus)、鞘氨醇单胞菌属(Sphingomonas)(图4)。其中CLU、ZBC 方法鉴定到的丰度最高菌属均为短螺旋体属(Brevinema),分别占样品的10.61%和9.15%,这个菌属在QIA 中仅为2.48%。QIA 方法提取的样品中丰度最高菌属为微小杆菌属(Exiguobacterium),在CLU 中仅为第四优势属。这表明在门、属水平上,三种不同DNA 提取方法导致了半滑舌鳎肠道菌群测序分析结果的差异。

图4 三种方法提取的DNA 样品中优势菌属相对丰度
2.4 Beta 多样性分析
基于UniFrac 距离计算样本间的距离矩阵来衡量三种样本间物种组成的相似度,发现CLU 和QIA 方法提取的样品之间的微生物群落结构和物种丰富度更相似(图5)。

图5 基于距离的加权UniFrac 分析三种DNA 样本相似性
3 讨论
3.1 影响DNA 提取效率的因素
高通量测序技术具有定量准确和测序深度高的优点,能够更真实地反映菌群结构特征,已广泛应用于分析复杂的肠道菌群结构[27]。高质量的DNA 提取是准确测序的前提,现已有多种提取肠道菌群基因组DNA 的方法,但提取效率各不相同[28]。无论何种方法,DNA 的提取都需要细胞裂解和核酸纯化两个步骤,通常是利用物理、化学或酶解作用裂解细胞,使DNA 释放出来,进一步纯化与裂解体系中其他杂质分离。不同提取方法可能对裂解效果具有明显差异,如何提高细胞裂解效率是肠道菌群多样性研究中关键问题。在DNA 提取过程中,不同的细菌类群(如革兰氏阴性菌、革兰氏阳性菌等)对使用的化学试剂表现出不同程度的抗性[29]。常用的化学试剂有十二烷基硫酸钠(SDS)和十六烷基三甲基溴化铵(CTAB)。SDS 是一种阴离子表面活性剂,可与膜脂蛋白作用从而破坏细胞膜结构;而CTAB 是一种阳离子去污剂,能够溶解细胞膜并与核酸形成复合物[30]。化学法具有成本低、DNA 提取产量高、降解少的优点[31]。但化学试剂并不能稳定地破坏细菌细胞壁,可能会阻碍DNA 释放或在裂解过程中破坏DNA,这些因素导致微生物群落中不同细菌类群的低估或高估。以往研究表明,溶菌酶可以选择性分解微生物细胞壁而不破坏其他成分[32],但溶菌酶对革兰氏阴性菌DNA 提取效率不高[33]。为了解决这一问题,本实验室利用CLU 方法将溶菌酶和超声波裂解法结合以提取不同类群细菌基因组DNA。在本研究中采用CLU 方法提取的半滑舌鳎肠道菌群DNA 经测序分析后,鉴定到较多的细菌类群。这表明使用适当的超声波破碎处理来裂解革兰氏阴性菌细胞壁,可以使DNA 有效地释放。此外,该方法中使用的水解酶、蛋白酶K 和RNase A 的协同作用也有助于DNA 释放和纯化。
3.2 不同DNA 提取方法引起菌群结构分析结果的差异
迄今,对于鱼类肠道菌群基因组DNA 的提取方法尚无统一的标准。本研究采用三种不同方法提取半滑舌鳎肠道菌群DNA,所得样品经过测定得到的OTU 只有不到50%是共有的。与QIA 样品相比,CLU 和ZBC 样品中测定得到的肠道菌群OTU 数、Alpha 多样性指数(Chao1 和Simpson)明显较高。
在门水平上,螺旋菌门(Spirochaetae)丰度在CLU 和ZBC 样品中分别为9.18%和10.73%,而在QIA 中仅为2.52%。在属水平上,短螺旋体属(Brevinema)的丰度在CLU 和ZBC 样品中分别为10.61%和9.15%,而在QIA 中仅为2.48%。不同细菌类群丰度的差异代表相应基因序列丰度不同,这可能是由于不同DNA 提取方法对肠道菌群DNA 的提取效率不同。本研究通过三种方法提取DNA,鉴定到的丰度较高的细菌门类是相同的,但对于低丰度的细菌门类不能完全鉴定到,在属的水平上也发现了相似情况。因此,同时选用多种不同方法提取DNA,用于高通量测序分析,最后得到的菌群组成信息更加全面、准确。
3.3 半滑舌鳎肠道菌群结构特征
三种方法提取的DNA样品经高通量测序分析发现,半滑舌鳎肠道菌群中丰度最高的菌门为变形菌门(Proteobacteria),其次是厚壁菌门(Firmicutes)、螺旋菌门(Spirochaetae)、拟杆菌门(Bacteroidetes),这与张正等[34]的研究结果相似,表明这些菌门可能是半滑舌鳎肠道内核心菌群。本研究发现半滑舌鳎肠道菌群中含有较高丰度的螺旋菌门(Spirochaetae),这与在锦鲤(Cyprinus carpiovar.Koi)[21]和异育银鲫(Carassius auratus gibelio)[35]中的研究结果不同。由此推测,不同的系统发育关系、饮食习性和生活环境等因素可能影响鱼类肠道菌群的结构组成。另外,在患病半滑舌鳎中螺旋菌门(Spirochaetae)丰富度明显降低[36],这表明螺旋菌门(Spirochaetae)的丰富度可能与半滑舌鳎健康状况密切相关。
本研究表明,不同方法提取的DNA 经高通量测序分析鉴定到的主要优势菌群类别是一致的,但对一些丰度较低的菌群类别鉴定的结果存在偏差。此外,本研究揭示了健康半滑舌鳎肠道核心菌群主要包括变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、螺旋菌门(Spirochaetae)、拟杆菌门(Bacteroidetes)。研究结果为半滑舌鳎肠道菌群多样性研究方法,特别是DNA 提取策略提供了参考资料,同时有助于全面了解健康半滑舌鳎的肠道菌群结构特征。
猜你喜欢
天津农学院学报(2024年1期)2024-04-07 11:20:22
水产学报(2024年3期)2024-03-25 07:43:22
国际太空(2023年1期)2023-02-27 09:03:42
透析与人工器官(2020年1期)2020-11-16 01:42:34
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
铁道通信信号(2019年8期)2019-10-10 05:06:00
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
中国发展观察(2017年8期)2017-04-26 03:51:50
