
植物靛蓝的质量分析与评价
2022-09-27 08:06:10柳成航徐增莱李冬玲郑玉红储力群
植物资源与环境学报 2022年5期
柳成航,徐增莱,李冬玲,郑玉红,汪 琼,①,储力群
〔1.江苏省中国科学院植物研究所(南京中山植物园),江苏 南京 210014;2.常州云卿生物科技有限公司,江苏 常州 213004〕
随着人们环保意识的增强,植物染料因无毒无害、环保绿色的优点再次回到人们的视野中,尤其是植物靛蓝的研究和应用逐渐丰富起来。靛蓝是一种历史悠久的蓝色染料,与常用中药青黛为同一植物来源,收载于《中国药典》[1]。在中国贵州和云南等省的山村地区,产靛植物的种植以及制靛染色行业已成为当地的特色产业[2]。然而,当前中国植物靛蓝行业仍处于传统的粗放生产经营状态,往往以农户为经营单位,种植规模小,制靛技术水平较低,产品质量良莠不齐。靛蓝质量全凭主观经验判断,其真实含量无法有效检测,产品质量不能与价格相关联,严重制约行业的健康发展。
近年来,靛蓝含量的检测方法包括高效液相色谱(HPLC)法[3,4]、液质联用法[5]和定量核磁共振氢谱法[6]等,其中广泛使用的HPLC法具有测量准确、分离度好、消耗样品量少等优点,但也存在着测试成本高昂、溶剂消耗量大、测试时间偏长、设备价格较高等问题。由于产靛植物种植地区往往经济不发达,直接面向种植与靛蓝初加工的企业均为涉农小微企业,经营规模较小,技术水平较低,因此,开发一种廉价快速、高效准确的检测植物靛蓝样品中靛蓝含量的方法很有必要。
本研究使用分光光度法对收集的32个靛蓝样品进行了靛蓝含量测定,将该方法与HPLC法和高锰酸钾滴定法进行对比,并对32个样品中的靛蓝含量及其与产靛植物的种类和制备工艺等因子的关系进行了分析。以期提供一种操作简便、成本低廉、结果准确的靛蓝含量测定和评价方法,进一步提升山村地区植物靛蓝行业产品质量,推动行业良性健康发展。
1 材料和方法
1.1 材料和仪器
于2018年至2021年收集植物靛蓝样品,样品的详细信息见表1。样品为膏状半固体,冷冻干燥后粉碎过40目筛,置于避光阴凉处保存、备用。
主要仪器:UV-8000紫外-可见分光光度计(上海元析仪器有限公司)和Agilent1100高效液相色谱仪(美国Agilent公司)。主要试剂:靛蓝标准品(纯度99.6%,上海诗丹德标准技术服务有限公司)、甲醇(色谱纯,美国Tedia公司)、N,N-二甲基甲酰胺(DMF,分析纯,广东光华科技股份有限公司)、2,6-二叔丁基对甲基苯酚(BHT,分析纯,国药集团)。
1.2 方法
1.2.1 分光光度法标准曲线绘制 精密称取1.1 mg靛蓝标准品,置于50 mL容量瓶。用含质量体积分数0.5%BHT的DMF溶液溶解并定容,得到22.00 mg·L-1的靛蓝标准品母液。将配制好的靛蓝标准品母液用含质量体积分数0.5%BHT的DMF溶液稀释,得到1.38~14.67 mg·L-1的系列靛蓝溶液。以含质量体积分数0.5%BHT的DMF溶液作为空白对照,于波长610 nm处测定不同浓度的靛蓝溶液的吸光度。以吸光度为纵坐标(y)、靛蓝浓度为横坐标(x)绘制标准曲线。回归方程为y=0.077 54x+0.013 08(r2=0.999 6),在1.38~14.67 mg·L-1范围内线性关系良好。
1.2.2 样品溶液配制及检测分析 称取靛蓝样品约5 mg,置于25 mL容量瓶。加入15 mL含质量体积分数0.5%BHT的DMF溶液,室温下超声(300 W,35 kHz)5 min促进溶解,冷却后用含质量体积分数0.5%BHT的DMF溶液定容。重复取样,同一样品配置3个样品溶液,按上述方法配制样品溶液。分析前用尼龙微孔滤膜(孔径0.45 μm)过滤。以含质量体积分数0.5%BHT的DMF溶液为空白对照,于波长610 nm处测定样品溶液的吸光度,根据标准曲线计算样品溶液浓度并计算样品中的靛蓝含量。
1.2.3 HPLC法和高锰酸钾滴定法测定靛蓝含量 参考董娟娥等[7]报道的色谱条件和分析方法对M1、M2、M3、M4、M12、M14、M16、M25和M27 9个植物靛蓝样品中的靛蓝含量进行测定。参考化工行业标准HG/T 2750—2012《靛蓝》中描述的高锰酸钾滴定法,对植物靛蓝样品M14的靛蓝含量进行测定。
1.3 数据统计
采用EXCEL 2019软件对相关数据进行统计和计算。
2 结果和分析
2.1 样品中靛蓝含量的比较
分光光度法的测定结果(表1)表明:32个样品中的靛蓝含量为0.34%~8.69%,均值为3.95%。靛蓝含量最高的样品为贵州三都的M25,含量最低的样品为四川凉山的M1,前者为后者的25.6倍。湿法工艺下,8个来源于蓼蓝〔Persicariatinctoria(Aiton)Spach〕的样品M3至M10中靛蓝含量为1.10%~7.99%,均值为4.05%;17个来源于板蓝〔Strobilanthescusia(Nees)Kuntze〕的样品M16至M32中靛蓝含量为1.69%~8.69%,均值为4.16%;2个来源于菘蓝(IsatistinctoriaLinn.)的样品M11和M12中靛蓝含量分别为1.93%和4.95%,均值为3.44%;2个来源于野青树(IndigoferasuffruticosaMill.)的样品M14和M15中靛蓝含量分别为3.43%和7.31%,均值为5.37%;1个来源于木蓝(IndigoferatinctoriaLinn.)的样品M13中靛蓝含量为4.65%。
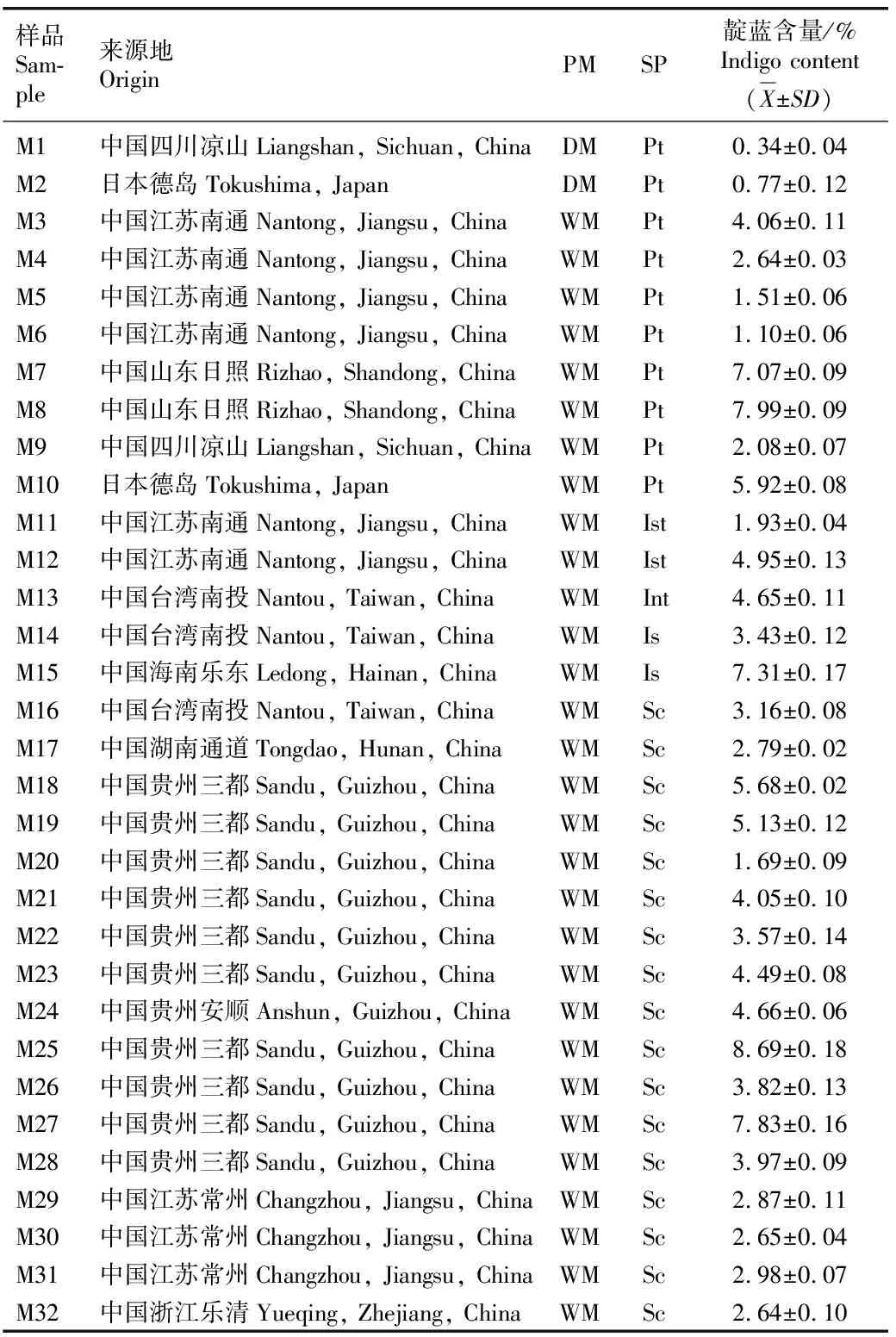
表1 采用分光光度法测定不同来源的植物靛蓝样品中的靛蓝含量(n=3)1)
2.2 与HPLC法和高锰酸钾滴定法的比较
HPLC法测定结果显示:四川凉山的样品M1,日本德岛的样品M2,江苏南通的样品M3、M4和M12,台湾南投的样品M14和M16,贵州三都的样品M25和M27中靛蓝含量分别为0.26%、0.53%、3.54%、2.61%、4.77%、2.97%、2.76%、8.33%和7.44%。HPLC法和分光光度法测得的靛蓝含量接近,分光光度法的测量结果比高效液相色谱法高0.03%~0.52%。高锰酸钾滴定法测定结果显示:样品M14中靛蓝含量为2.81%,测定的结果与HPLC法接近。
3 讨论和结论
溶液中和织物上的靛蓝会被氧气缓慢氧化,生成靛红、邻氨基苯甲酸和色胺酮等产物[8],导致靛蓝溶液或织物褪色。为了抑制靛蓝氧化,本研究在溶剂中加抗氧化剂BHT,少量BHT能有效抑制靛蓝氧化,增加靛蓝溶液稳定性[9]。
本文中,分光光度法测得的靛蓝含量比HPLC法的高0.03%~0.52%。这可能是由于分光光度法没有选择性,样品中的少量伴生成分靛玉红及其他杂质对波长610 nm处的吸光度有一定贡献,造成分光光度法的测量结果偏高。但分光光度法操作简单,仪器普及。高锰酸钾滴定法的测定结果尽管与HPLC法接近,但其相对标准差较大(SD=0.25%,RSD=8.88%,n=3)。这是由于天然靛蓝样品纯度不高,成分复杂[10],其中存在的靛玉红等色素成分影响高锰酸钾滴定终点的颜色变化,导致滴定终点突跃不明显,影响终点的判断。总体而言,高锰酸钾滴定法仅适用于纯度较高的合成靛蓝样品的含量测定。
植物靛蓝的制备工艺分为干法和湿法,干法是将收割后的产靛植物进行堆积发酵;湿法是将产靛植物体浸泡于水中任其发酵,发酵后加入石灰水并鼓入空气进行“打靛”[11]。本研究中采用干法制备的样品M1和M2颜色灰暗,靛蓝含量较低(低于1.0%)。而同样源自蓼蓝,采用湿法工艺得到的样品M3至M10的靛蓝含量均高于1.0%,说明制备工艺对植物靛蓝样品中的靛蓝含量有较大影响。此外,沉淀剂石灰的量也会影响靛蓝含量。本研究收集的样品M4、M3和M7在制靛时加入了相当于蓼蓝质量2.5%、2.0%和0.8%的石灰,发现加入石灰的量越多,靛蓝样品中的靛蓝含量越低。
植物靛蓝样品中靛蓝含量与来源植物种类有关。湿法工艺下,来源于蓼蓝、板蓝、菘蓝、野青树和木蓝的样品中靛蓝含量的均值分别为4.05%、4.16%、3.44%、5.37%和4.65%。不同植物靛蓝样品中靛蓝含量从高至低依次为野青树、木蓝、马蓝、蓼蓝、菘蓝。
综上所述,分光光度法测量快速、准确,可用于染料、药品等行业的快速检测。采用湿法工艺得到的样品中靛蓝含量明显高于干法工艺,样品中靛蓝含量与产靛植物种类有关。
致谢:江苏南通水色染坊、台湾南投自然色手作坊、贵州亘蓝母图民族布艺蜡染开发有限公司提供了部分样品,谨致谢忱!
猜你喜欢
山东冶金(2022年3期)2022-07-19 03:25:36
云南化工(2021年11期)2022-01-12 06:06:18
食品安全导刊(2021年20期)2021-11-28 00:56:56
供水技术(2021年3期)2021-08-13 09:08:36
宜春学院学报(2020年9期)2020-12-03 06:22:10
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30 12:26:34
现代园艺(2017年21期)2018-01-03 06:41:34
河北地质(2016年2期)2016-03-20 13:52:04
林业与生态(2016年2期)2016-02-27 14:23:55
中国卫生标准管理(2015年6期)2016-01-14 05:17:17
