
致癌超级增强子的形成与干预研究进展
2022-09-13 08:15:26袁婺洲李瑞可
生命科学研究 2022年3期
袁婺洲,李瑞可
(湖南师范大学生命科学学院心脏发育研究中心,中国湖南 长沙 410081)
国际癌症研究机构(International Agency for Research on Cancer,IARC)发布的全球最新癌症负担数据显示:2020年全球新发癌症病例1 929万例,癌症死亡病例996万例。可见,癌症依然是本世纪最可怕的疾病之一,给人们的生活带来巨大阴影和负担,因而癌症发生、转移的分子机制以及治疗和预后策略一直是研究者们孜孜不倦关注的重大课题。近年很多文献报道,超级增强子(super enhancer,SE)在癌症发生、细胞分化、免疫应答等重要生物学过程中发挥着重要调控作用[1~3];肿瘤细胞的许多关键致癌基因由超级增强子驱动,超级增强子有潜力成为理想的抗癌靶点[1,4]。
1 增强子与超级增强子的概念
1981年,Banerji等[5]在猿猴空泡病毒SV40 DNA的5'端上游发现了首个增强子,它能增强哺乳动物细胞中靶基因转录。随后,人们陆续发现许多增强子,它们都能通过转录因子募集共激活因子(例如中介体复合物CREB结合蛋白CBP和p300),从而改变染色质的空间结构,促进转录因子与增强子、启动子及RNA聚合酶相互作用[6]。增强子的活性也与组蛋白的修饰状态有关,因为基因活跃的转录起始位点都有H3K27me3、H3K27ac和H3K4me1标记[7~8]。此外,增强子还能被转录为增强子RNA(enhancer RNA,eRNA)[9]。
与普通增强子不同,超级增强子是一种跨越8 kb以上的大型增强子,能与细胞中多种组织特异性转录因子(如胚胎干细胞的Oct4、Sox2及Nanog等)、中介体复合物(mediator,Med)、染色质调节因子及RNA聚合酶Ⅱ复合物结合,且这些活性分子的密度是普通增强子的好几倍。因此,超级增强子比普通增强子具有更强的驱动靶基因转录的能力(图1A),且超级增强子产生的RNA(super enhancer RNA,seRNA)水平更高[1,4]。虽然超级增强子具有与普通增强子相似的作用机制,但在肿瘤发生过程中,DNA突变、插入或缺失、染色体重排、染色质三维结构改变和病毒感染则可能导致致癌超级增强子(carcinogenic super enhancer)的产生,促使癌细胞中癌基因高水平转录[10~21]。
2 超级增强子的结构和鉴定
超级增强子与普通增强子结构类似,都能与转录因子、中介体复合物、启动子、RNA聚合酶及靶基因形成一个环状结构,只是它比普通增强子更大(现倾向于认为至少跨越基因组8.7 kb大小),包含的增强子和转录因子更多[6](图1A)。中介体复合物、染色质因子、H3K27ac和H3K4me1、组蛋白乙酰基转移酶、p300和CBP、RNA聚合酶Ⅱ和eRNA等,大量富集在超级增强子区域,它们具有很强的染色质可及性[1]。此外,超级增强子在Wnt、转化生长因子-β(transforming growth factor-β,TGF-β)和白血病抑制因子(leukemia-inhibitory factor,LIF)信号转导途径中更受末端转录因子的束缚,调控基因表达水平更高[4,11,16,22~23]。一些证据表明,超级增强子中的组成增强子可能彼此加强或共同发挥作用,在基因调控中具有非冗余功能,而删除组成增强子则可能损害超级增强子其他成分的活性,导致整个超级增强子的功能障碍[23~25]。
研究人员提出了一种相分离模型来解释超级增强子的形成机制[26~31]。通过类似于聚合物缩合的相分离现象,蛋白质和DNA的异质混合物被组装成无膜细胞器结构[28]。其中,溴结构域蛋白4(bromodomain protein 4,BRD4)和Med 1(mediator 1)的内部无序区域(inner disorder,IDR)可能在超级增强子介导的转录位点形成相分离的液滴(图1B),促进特定基因转录成分的分隔和集中[29]。缺乏黏连蛋白会导致超级增强子在细胞核中广泛融合,从而限制超级增强子之间的相互作用[30~31]。

图1 超级增强子结构与相分离模型[31](A)增强子或超级增强子与转录调节因子结合,包括转录因子、共激活因子和RNA聚合酶Ⅱ复合体。黏连蛋白将增强子或超级增强子与转录调节因子黏连在一起形成一个环状结构。E表示增强子;TF表示转录因子;Med表示中介体复合物;RNA PolⅡ表示RNA聚合酶Ⅱ;(B)超级增强子激活的相分离模型。转录调节因子之间的高密度相互作用在超级增强子基因座处形成了相分离的多分子复合物,从而导致超级增强子驱动基因的转录。Fig.1 Structure of super enhancer and phase separation model[31](A)Enhancers or super enhancers bind to transcriptional regulators,including transcription factors,co-activators,and the RNA polymerase Ⅱ complex.Cohesin binds to enhancers or super enhancers and forms a loop with transcriptional regulators.E:Enhancer,TF:Transcription factor,Med:Mediator,RNA polⅡ:RNA polymerase Ⅱ;(B)A phase-separation model of super enhancer activation.High-density interactions among transcriptional regulators form phase-separated multimolecular complexes at super enhancer loci,resulting in super enhancer-driven gene transcription.
如何鉴定超级增强子呢?Young等[4,32~34]提出一个三步鉴定超级增强子的方法。第一步,运用染色质免疫沉淀测序(chromatin immunoprecipitation sequencing,ChIP-seq)获得小鼠胚胎干细胞DNA序列上Oct4、Sox2和Nanog等转录因子结合的信号强度,其中转录因子与增强子和转录起始位点密集结合的位点被用作超级增强子的候选片段[4]。第二步,在候选片段中选出共定位于12.5 kb DNA片段内的增强子,将其定义为复合增强子(stitched enhancer)。他们一共筛选出8 794个复合增强子。第三步,确定超级增强子和普通增强子之间的阈值。首先,将复合增强子和单个增强子按照ChIP-seq所测Med1信号水平的强度排序,绘制一张曲线图;随后,将曲线上斜率为1的切线点所得的Med1信号值设定为区分超级增强子和普通增强子的阈值,高于该阈值为超级增强子,低于该阈值为普通增强子。实验结果显示,在8 794个复合增强子中,一共有231个DNA片段的Med1信号值斜率大于1,这231个复合增强子片段即被鉴定为超级增强子[32~34](图2)。
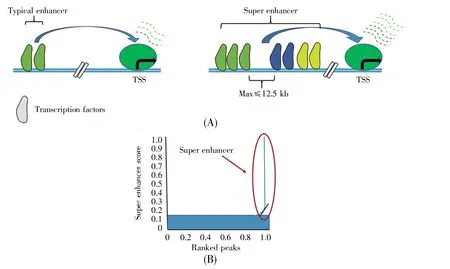
图2 普通增强子与超级增强子示意图[32](A)增强子是位于转录起始位点远端的具有方向和位置独立性的顺式调控元件[33~34]。共定位于12.5 kb DNA片段之内且Med1信号值大于阈值的增强子被称为超级增强子。超级增强子通常比普通增强子大一个数量级,具有更高的转录因子密度和更强的转录激活能力。TSS表示转录起始位点;(B)根据ChIP-seq信号水平的强度对增强子进行排序,确定阈值从而区分普通增强子和超级增强子。Fig.2 Schematic diagram of typical enhancers and super enhancers[32](A)Enhancers are orientation-and position-independent cis-regulatory elements distal to the transcription start site[33~34].Enhancers that co-localize within the 12.5 kb DNA fragment and their Med1 signal values are greater than threshold are called super enhancers.Super enhancers are usually an order of magnitude larger than ordinary enhancers,with higher density of transcription factors and stronger transcriptional activation capacity.TSS:Transcription start site;(B)Enhancers are ranked according to the intensity of ChIP-seq signal levels,and thresholds are determined to distinguish typical enhancers from super enhancers.
3 致癌超级增强子的发现
肿瘤发生时,肿瘤细胞可能获得特定的超级增强子来促进癌基因表达,介导信号通路失调,这种特定的超级增强子被称为致癌超级增强子[1,11,17,35]。致癌超级增强子首先发现于多发性骨髓瘤细胞(multiple myeloma cell),具有与Med1和BRD4富集结合的特点。根据H3K27ac ChIP-seq的数据,在直肠癌、前列腺癌及胰腺癌等18种人类癌症细胞中人们也鉴定到了超级增强子[7];后来又在神经母细胞瘤、髓母细胞瘤、食道癌、胃癌等发现超级增强子[36],说明肿瘤中普遍存在致癌超级增强子。
致癌超级增强子通过增加癌基因转录促使细胞向恶性肿瘤发展[23]。例如,致癌超级增强子可能激活MAPK信号通路,从而抑制细胞凋亡并增加细胞增殖[37];或者介导成红细胞病毒E26癌基因同源物ERG过表达,促进癌症发生[38]。另外,致癌超级增强子增加CYP24A1(1,25-dihydroxyvitamin D 24-hydroxylase)、GJA5(gap junction proteinα5)、SLAMF7(signaling lymphocyte activation marker family member 7)和 ETV1(E twenty-six variant 1)的表达[39]。相关研究报道,超级增强子增加了腺样囊性癌(adenoid cystic carcinoma,ACC)中端粒酶逆转录酶基因(telomerasereversetranscriptase,TERT)的表达[40];结直肠癌(colorectal cancer,CRC)相关的超级增强子在TCF4结合位点富集[23]。TCF4是Wnt信号转导的靶标,与c-MYC基因座结合,在癌细胞获得致癌超级增强子后显示出强H3-K27Ac信号[23]。针对MCF-7细胞中H3K27Ac的ChIP-seq分析表明,超级增强子靶向的ESR1(estrogenreceptor1)基因仅编码雌激素受体α(estrogen receptor,ERα)。在ER阳性乳腺癌细胞中,超级增强子靶向基因富集并与ERα结合,而在三阴性乳腺癌细胞中,超级增强子富集位点与致癌转录因子位点不同[41]。
4 致癌超级增强子的形成机制
大量的全基因组研究表明,与疾病相关的体细胞变异主要发生在非编码基因组中,且经常在基因调节区域富集[42~43]。生殖细胞和体细胞似乎通过多种机制获得超级增强子,包括基因组缺失、重复、易位、插入、倒位和单核苷酸多态性(single nucleotide polymorphism,SNP)等。这些DNA序列改变可以破坏特定超级增强子中的转录因子结合位点,改变超级增强子的拷贝数,并改变基因组构像,从而导致超级增强子的激活或抑制,最终导致超级增强子附近靶基因的失控[15,19]。除了基因突变和基因组改变,染色体重排、3D染色质结构变化和病毒感染等也能导致超级增强子形成[13~14,17,20-22,31,35,44~51]。
4.1 DNA序列改变形成致癌超级增强子
超级增强子的DNA序列突变能改变启动子和增强子的功能。例如,在T细胞急性淋巴细胞白血病(T-cell acute lymphoblastic leukemia,T-ALL)中,TAL1(T-cellacutelymphocyticleukemiaprotein1)基因上游非编码区域的2~18 bp小片段插入产生了转录因子MYB的从头结合位点,导致超级增强子形成并驱动TAL1表达。MYB募集CBP/p300乙酰转移酶和TAL1转录因子复合物,进一步驱动白血病关键基因表达(图3A)。除了小片段插入外,SNP也经常被发现可以启动致癌超级增强子的活性。例如:在神经细胞瘤(neuroblastoma)中,LMO1(LIMdomainonly1)癌基因座超级增强子的形成取决于GATA3与保守GATA位点的结合。位于超级增强子附近的SNP改变了保守的GATA结合位点,将GATA变为TATA位点,导致超级增强子活性和LMO1表达显著降低[46]。另外,SNP破坏与肿瘤抑制基因相关的超级增强子,从而促进肿瘤发生。全基因组关联分析显示,BMF(BCL2-modifyingfactor)基因的15q15.1风险基因座具有慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)易感性。15q15.1风险基因座中的SNP产生超级增强子,以调节促凋亡基因BMF,并破坏核因子-κB(nuclear factor-κB,NF-κB)与超级增强子结合,从而增强BCL2抗凋亡功能,促进肿瘤发生[45](图 3B)。
4.2 染色体重排形成致癌超级增强子
染色体重排、倒位、易位和缺失使超级增强子从其天然基因区域转移至癌基因区域,从而激活癌基因。这种现象被称为“超级增强子劫持”,并已在多种癌症中得到报道,包括急性髓细胞白血病(acute myeloid leukemia,AML)、神经母细胞瘤、髓母细胞瘤和结直肠癌[13,15,20,35,47]。最经典的例子是,AML细胞中一个9 kb片段的倒位,引起超级增强子从原来的GATA2肿瘤抑制因子重新定向到EVI1(ecotropicvirusintegration-1)癌基因增强子,从而导致肿瘤抑制因子的下调和癌基因激活[13]。增强子劫持的另一个例子是,在腺样囊性癌中一个染色体易位将MYB基因远端超级增强子重新定位到MYB基因的近端,导致MYB高表达[49]。进一步的染色体构象捕获(chromosome conformation capture,3C)分析证实,MYB启动子和异常转移的超级增强子之间的染色质相互作用[52],而且易位的超级增强子元件包含MYB结合位点,可被MYB自身主动结合并形成正反馈环,从而进一步增强MYB表达。新近发现的一例增强子劫持是C19MC-TTYH1(chr19q13.41miRNAcluster-Tweetyhomologue1)基因融合产生的复合超级增强子,其扩增了C19MC-LIN28A-MYCN(chr19q13.41miRNAcluster-Lin28homologA-MYCN)致癌回路,并驱动了DNA甲基转移酶3B6(DNMT3B6)的表达,从而促进了胚胎肿瘤的恶性表观遗传状态[53]。除基因组重排外,拷贝数变异也可以导致致癌超级增强子激活。体细胞拷贝数和12种癌细胞的组织特异性表观遗传分析表明,KLF5(krüppellikefactor5)、USP12(ubiquitin-specificprotease12)、PARD6B(par-6familycellpolarityregulatorbeta)、MYC和其他癌症相关基因附近超级增强子的局部扩增,可以驱动癌基因的异常表达[48]。
4.3 3D染色质结构变化形成致癌超级增强子
哺乳动物基因组被划分为一系列平均大小约为1 Mb的拓扑关联域(topologically associating domain,TAD)。这些TAD是染色体的结构和功能单元,不同细胞类型中TAD结构是保守的[30,54]。TAD具有限制长距离增强子与启动子相互作用的功能,从而使启动子与远端增强子和超级增强子隔离[12,19,54](图 3C)。

图3 致癌超级增强子形成的各种机制[31](A)在TAL1癌基因上游非编码区域的小片段插入诱导了转录因子MYB的从头结合位点,导致驱动TAL1表达的超级增强子形成。MYB结合并募集其H3K27乙酰化酶结合伴侣CBP,即含有RUNX1和GATA3的TAL1转录复合物;(B)15q15.1风险基因座中的SNP产生促凋亡基因BMF的超级增强子,并破坏转录因子RELA与超级增强子的结合,从而激活BCL2的抗凋亡功能,促进肿瘤发生;(C)致癌基因的激活通过结构变异或表观遗传效应改变而发生。Fig.3 Various mechanisms of carcinogenic super enhancer formation[31](A)Insertion of a small fragment in a noncoding region upstream of the TAL1 oncogene induces a de novo binding site for the transcription factor MYB,resulting in the formation of a super enhancer that drives TAL1 expression.MYB binds and recruits its H3K27 acetylase-binding partner CBP,the TAL1 transcription complex containing RUNX1 and GATA3;(B)SNPs in the 15q15.1 risk locus generate a super enhancer of the pro-apoptotic gene BMF and disrupt the binding of transcription factor RELA to the super enhancer,thereby activating the anti-apoptotic function of BCL2 and promoting tumorigenesis;(C)Activation of oncogenes occurs through structural variation or altered epigenetic effects.
TAD由染色质环组成,通常涉及CTCF(CCCTC-binding factor)和黏连蛋白。与CTCF相关的边界元件的存在可防止TAD异位接触,并使TAD与邻近增强子隔离。近期,研究人员开发了一种顺式表达结构改变作图算法,用于系统预测癌症相关基因过度表达的概率。研究者们通过这种方法发现了TAD边界缺失事件,该事件导致活性染色质扩散到相邻融合的TAD并产生超级增强子元件,从而增加IRS4(insulinreceptorsubstrate4)基因在肉瘤(sarcoma)和鳞状癌(squamous cancer)细胞中的表达[47]。此外,研究人员通过对CTCF和黏附素结合位点的分析获得了多种癌细胞类型的突变。例如,CTCF和黏附素单倍体不足的小鼠易患癌症[55]。在T细胞急性淋巴细胞白血病细胞中,CTCF结合位点破坏TAD边界的调控元件,激活TAL1和LMO2表达,从而导致T细胞转化[56]。除了TAD边界的突变外,表观遗传调控的改变也已被证明是胶质瘤(gliomas)中TAD破坏的机制[57]。据报道,CTCF位点甲基化的增强和CTCF结合力的降低会引起TAD结构的部分破坏,从而导致PDGFRA(platelet-derivedgrowthfactorreceptoralpha)基因的活化[57]。
4.4 病毒感染形成致癌超级增强子
病毒感染也能诱导超级增强子形成,从而驱动与细胞增殖和存活相关的关键基因的高水平转录。具有致癌活性的病毒包括EB病毒(Epstein-Barr virus,EBV)、人乳头瘤病毒(human papilloma virus,HPV)、人T细胞白血病病毒(human T cell leukemia virus,HTLV)和乙型肝炎病毒(hepatitis B virus,HBV)。EBV感染人类B细胞后,会产生癌蛋白,包括EBNA2(Epstein-Barr virus-encoded nuclear antigen 2)、EBNA3A、EBNA3C 和 EBNALP(Epstein-Barr virus-encoded nuclear antigen leader protein)。这些癌蛋白激活NF-κB亚基并与超级增强子结合,以驱动一些癌基因和抗凋亡基因表达,包括MYC、miR155、IKZF3和BCL2,从而促进淋巴母细胞生长[14,21]。此外,EBV超级增强子(EBV super-enhancer,ESE)能被转录为seRNA,促进MYC癌基因的转录激活[50]。Ⅰ型HTLV(HTLV-1)经常引发成人T细胞白血病/淋巴瘤(adult T-cell leukemia/lymphoma,ATLL)。ATLL细胞的增殖依赖于BATF3(basic leucine zipper ATF-like transcription factor 3)和 IRF4(interferon regulatory factor 4)的表达,它们共同驱动ATLL特异性基因表达。病毒转录因子HBZ(HTLV-1 basic leucine zipper factor)在所有ATLL病例中表达,且HBZ与BATF3超级增强子结合调节BATF3和MYC基因的表达,从而促进ATLL细胞增殖[58]。
5 致癌超级增强子涉及的信号通路
致癌超级增强子富含与癌症信号通路相关的转录因子结合位点,通过调节靶基因激活包括Wnt、TGF-β和LIF在内的几种信号通路[2,23,59~61]。
致癌超级增强子介导的Wnt通路在肿瘤发生中起重要作用。先前的研究表明,在结直肠癌细胞中,与Wnt通路相关的超级增强子富含TCF4结合位点[23]。在基底细胞癌(basal cell carcinoma,BCC)的小鼠模型中,细胞是维持干细胞特性还是分化为特种细胞,与染色质状态改变、Wnt信号的快速激活以及超级增强子的重新编程都有关系。用Wnt信号抑制剂治疗BCC可减少残留的肿瘤负荷并增强细胞分化[59]。近期研究表明,Wnt信号和AHCTF1(AT-hook containing transcription factor 1)通过超级增强子介导的基因促进致癌基因MYC表达[60]。MYC的癌细胞特异性门控由AHCTF1调控,它通过β-catenin将核孔蛋白与致癌超级增强子连接起来[60]。
此外,TGF-β和LIF信号在癌症的发生中也起着至关重要的作用。在胰腺癌(pancreatic cancer)细胞中,TGF-Ⅱ型受体(TGF beta typeⅡreceptor,TGFBR2)中超级增强子的缺失显著下调了TGFBR2的表达,导致TGF-β诱导癌细胞的迁移和上皮间质转化(epithelial mesenchymal transition,EMT)受损[2]。用LIF重组蛋白处理的骨肉瘤(osteosarcoma)细胞显示出致癌细胞的迁移速率增加。X染色体上的UTX(ubiquitouslytranscribedtetratricopeptiderepeat)基因是LIF转录的关键激活因子。UTX抑制剂GSK-J4损害LIF基因位点的超级增强子,导致LIF信号通路受到抑制[61]。
除了上述信号通路外,其他一些信号途径也与致癌超级增强子有重要的关系。例如,突变的RAS活性促进致癌超级增强子形成,抑制RAS信号将导致超级增强子失活,同时降低癌基因表达[62]。
6 针对超级增强子靶标的癌症治疗策略
既然超级增强子能引起相关癌基因转录水平提高,那么,阻断超级增强子应该是一种可行的抗癌治疗策略。图4展示了两种通过靶向与超级增强子相关的复合物来抑制癌基因表达的潜在癌症治疗策略。
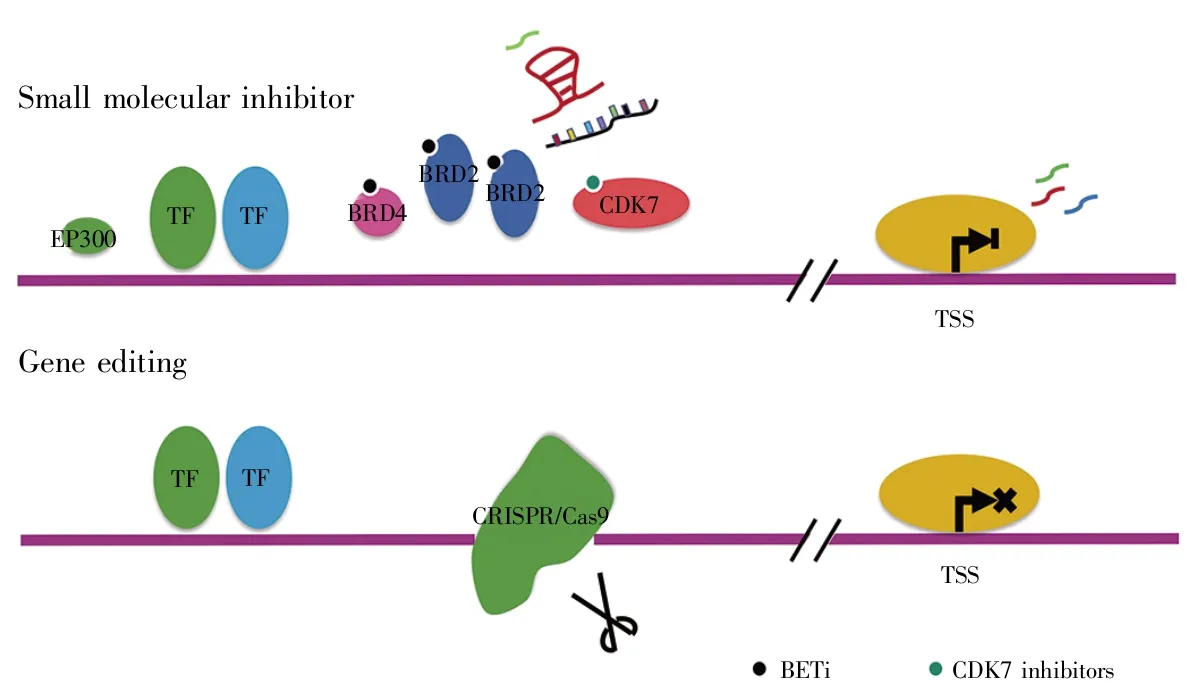
图4 靶向超级增强子复合物的两种方法示意图[95]小分子抑制剂和基因编辑可以靶向超级增强子相关复合物(例如BRD4和CDK7),从而破坏超级增强子功能。Fig.4 Schematic diagram of two methods for targeting super enhancer complexes[95]Small molecular inhibitors and gene editing can target super enhancer associated complexes such as BRD4 and CDK7,thereby disrupting super enhancer function.
6.1 小分子抑制剂
目前,设计用于癌症治疗的小分子抑制剂包括周期蛋白依赖性激酶7(cyclin-dependent kinase 7,CDK7)抑制剂(如 THZ1)、BRD4 抑制剂(如 JQ1或BETi)及其他抑制剂3种类型[11,16,22,37,49,63~95](表1)。
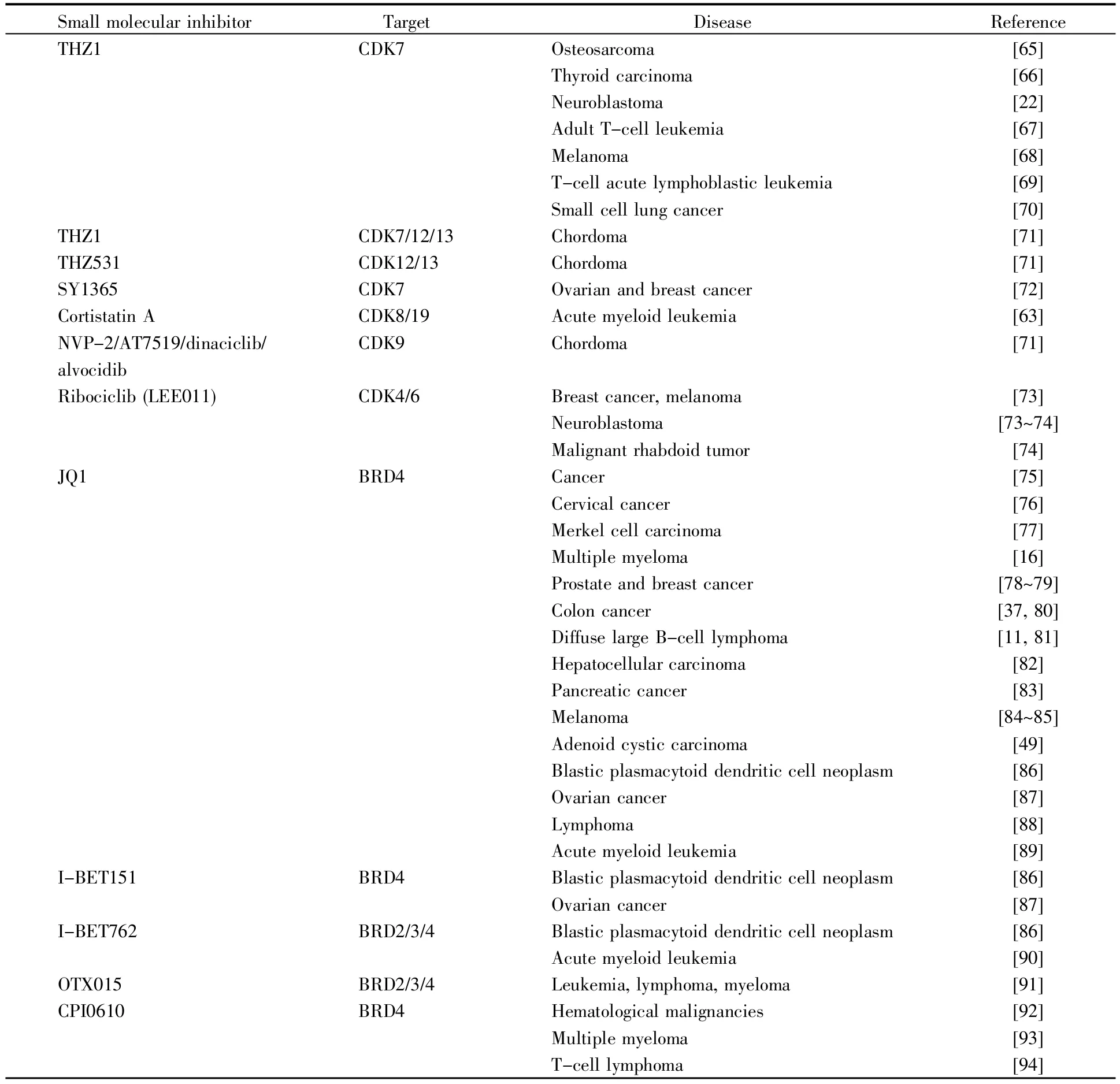
表1 致癌超级增强子的小分子抑制剂Table 1 Small molecular inhibitors of carcinogenic super enhancers
CDK属于丝氨酸/苏氨酸激酶家族,在调节细胞周期和癌基因转录过程中起着至关重要的作用[96]。据报道,CDK7不仅影响细胞周期,还与超级增强子驱动的癌基因调控有关,因此CDK7已成为有吸引力的抗癌靶标[72,97]。THZ1是最有效的CDK小分子抑制剂(CDK7i)之一,在多种肿瘤中具有抗癌作用,这主要归因于它对癌基因转录或超级增强子功能的干扰[97~98]。不过,研究表明THZ1也能抑制肌肉细胞的分化,提示THZ1在治疗癌症过程中可能对肌肉功能产生副作用[99]。CDK7i也可以与其他抗肿瘤药物联用,以提高疗效并减少副作用。例如,在弥漫性桥脑神经胶质瘤(diffuse intrinsic pontine glioma)中,CDK7i与组蛋白脱乙酰基酶抑制剂联用时,癌细胞对CDK7i的敏感性显著提高[97]。
BRD家族由4个成员组成:BRD2、BRD3、BRD4和BRDT,它们可以识别组蛋白乙酰化并促进相应癌基因的表达[100~101]。BETi(bromodomain and extraterminal domain inhibitor)主要抑制中介体复合物BRD4与超级增强子的结合,从而抑制超级增强子驱动的癌基因表达并减弱癌细胞的增殖[102]。不过,BETi抑制MYC基因的表达存在争议的报道。一方面,一些研究发现BETi会优先影响超级增强子驱动的MYC癌基因在多发性骨髓瘤和结肠癌中的表达,表明BETi敏感性与c-MYC基因水平显著相关[16,80];另一方面,也有研究没有观察到结肠癌中JQ1(BETi的一种)敏感性与c-MYC表达之间的显著相关性[37]。这说明针对患者需要量身定制个性化的治疗方法[83]。目前,多个Ⅰ期临床试验报告,BETi具有严重的副作用,包括心脏毒性、胃肠道毒性、贫血、腹泻、疲劳、恶心、中性粒细胞减少和血小板减少症等[91,103~104]。但是,BETi与维生素C的组合使用,可以在很大程度上减轻这些副作用[105]。
除了上述两类抑制剂外,一些基因表达的小分子蛋白质或表观遗传修饰也参与了阻断超级增强子的功能。例如:破坏KDM5C[lysine(K)-specifichistonedemethylase5C)基因与超级增强子形成的甲基化修饰(H3K4me1和H3K4me3),为乳腺癌治疗提供了一种新方法[106];在B细胞系急性淋巴细胞白血病中过表达IKAROS基因,可以干扰MYC超级增强子区域的H3K27ac3修饰,从而抑制MYC基因的表达[107];PAX3和PAX5是超级增强子在肺泡肺横纹肌肉瘤(pulmonary alveolar rhabdomyosarcoma)和慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)中的关键转录因子,PAX抑制剂具有显著的抗肿瘤作用[108~109]。此外,在脊索瘤(chordoma)中,IRS4/IGF2蛋白可以直接与超级增强子序列相互作用,抑制超级增强子的功能[71]。
6.2 基因编辑
癌细胞中异常的碱基插入、缺失和染色质重排可导致超级增强子形成。CRISPR/Cas9基因编辑技术可用于消除上述原因引起的超级增强子的形成,因此我们可以通过CRISPR/Cas9系统实现抗癌治疗。例如,RUNX1(RUNX family transcription factor 1)是调节正常和恶性血细胞产生的转录因子,通过CRISPR/Cas9系统破坏RUNX1基因的超级增强子区域会增加急性白血病细胞的凋亡,进而改变AML小鼠的存活率[110]。在T细胞急性淋巴细胞白血病的原始样本和细胞系中,TAL1基因转录起始位点上游7.5 kb处发生插入/缺失突变,促使MYB结合位点和超级增强子形成,从而导致癌基因表达上调;利用CRISPR/Cas9系统敲除异常插入的碱基后,急性淋巴细胞白血病样本中不再有超级增强子和TAL1基因过表达的现象[17]。此外,CRISPR/Cas9敲除系统也可用于消除AML细胞中异常增强子的激活。
CRISPR/Cas9敲入系统在与SNP相关的超级增强子驱动癌基因的表达模式中也很有前景[111~112]。在结肠细胞中,SNP rs6854845在超级增强子区域形成并影响超级增强子区域中H3K4me1和H3K27ac的转移富集,进而影响超级增强子驱动基因的表达,因此,SNP rs6854845被认为是结肠癌的危险因素之一[113~114]。基于CRISPR/Cas9的SNP位点基因编辑可以通过逆转SNP和超级增强子驱动基因之间的相互作用来改善细胞中的多种致病性变化。
7 总结与展望
致癌超级增强子具有比普通增强子更强的激活癌基因转录的能力,因而是很多恶性肿瘤发病的重要原因。致癌超级增强子的形成主要是由于癌基因增强子调控区域的DNA序列突变、碱基缺失或插入、染色体结构重排、染色质结构TAD边界缺失以及某些病毒感染,其导致转录因子和中介体复合物与普通增强子及启动子形成强大的超级增强子结构并被异常激活,随后通过影响Wnt、TGF-β、LIF及Ras等信号通路促使癌基因、抗凋亡基因或抑癌基因异常表达,最终促进肿瘤发生。虽然迄今针对致癌超级增强子的精细结构和致病机理的研究还不够深入,但是已知的致癌超级增强子的形成机制,还是能为肿瘤预防和治疗提供新的思路。例如:发生在增强子区域的有害序列变异可以通过广泛使用的CRISPR基因编辑工具进行修复;转录因子和中介体复合物的异常激活可以通过一些小分子抑制剂实现靶向抑制。以往有关抗肿瘤药物的设计主要关注癌基因或抑癌基因突变及其异常激活,但耐药性的存在及癌基因的低突变频率是不可忽视的影响因素[115~116]。近年来,随着表观基因组学研究的深入及致癌超级增强子的不断发现,基于表观遗传修饰设计的药物以及针对超级增强子及其相关复合物的小分子抑制剂和基因编辑疗法,可能具有更好的抗肿瘤应用前景[117]。
猜你喜欢
畜牧兽医学报(2022年3期)2022-03-30 02:29:20
中国畜牧兽医(2022年1期)2022-02-15 10:46:40
畜牧兽医学报(2021年12期)2021-12-31 01:38:10
生命科学研究(2020年6期)2021-01-09 11:34:24
现代泌尿外科杂志(2019年10期)2019-10-31 07:31:54
生物学通报(2019年2期)2019-06-15 01:33:42
遗传(2019年1期)2019-01-30 06:39:30
生物技术通讯(2017年4期)2017-11-06 01:24:56
健康管理(2016年2期)2016-05-30 21:36:03
中国医疗美容(2015年1期)2015-07-12 10:06:52
