
电化学氮气还原催化剂设计方法及研究进展
2022-09-09 15:44于丰收田李园
河北工业大学学报 2022年4期
于丰收,田李园
(河北工业大学 化工学院,天津 300131)
0 引言
氨气(NH3)作为一种基础化学品在化工、农业等领域有着广泛应用[1-2]。含NH3化合物为植物生长提供足够的氮肥,缓解地球有限的耕地资源与庞大的粮食需求矛盾。自人类掌握合成NH3工艺以来,世界人口数量由最初的不到2亿增加到了现在的56亿。此外,NH3含有17.6%(质量分数)的H,是一种重要的氢载体,与气态H2相比更容易储存和运输[3]。作为无碳储能中间体,NH3完全燃烧产物为N2和H2O,不会造成CO等有害气体和CO2等温室气体以及其他酸性气体的排放[4-5]。由于以上诸多优势,氨的需求量十分巨大,2018年全球共生产约1.7亿t氨,随着工、农业发展和人口增长,NH3需求量还会继续增加。目前,工业上主要采用Haber-Bosch工艺,在高温(300~600 ℃)和高压(20~40 MPa)条件下,以H2和N2为原料合成NH3[6-7]。作为线性非极性分子,N2的活化需要高能量输入如高温促进N≡N三键断裂。然而Haber-Bosch工艺合成NH3是放热过程,高温不利于热力学平衡向NH3生成方向移动。因此为了协调动力学与热力学限制,提高NH3生成率,Haber-Bosch 工艺选择在高温、高压条件下,经过多次循环提高反应转化率。经过多次转化,Haber-Bosch工艺合成NH3转化效率可达95%。NH3的需求量大,生产过程严苛,使合成氨工业成为目前能耗最高的化工过程,每年消耗的化石能源占全球总能耗的1%~2%[8]。作为原料之一,H2主要来源天然气重整。该过程消耗的天然气占全球总天然气消耗量的3%~5%,并造成每年数百万吨CO2排放。考虑到Haber-Bosch工艺高能耗、高排放等问题,开发替代工艺以更加绿色、可持续的方法实现NH3的高效合成迫在眉睫[9]。
近年来,电化学合成方法在H2O分解、CO2还原、O2还原等研究领域取得飞速发展[10-11],该绿色过程也被拓展到了NH3合成领域。电化学N2还原是在常温、常压下,以H2O为质子和电子源,在外加偏压条件下直接将N2还原成NH3[12]。所需的电能可以是可再生风电、太阳能发电或富余核电等洁净电能。电催化合成NH3过程兼具反应条件温和、设施要求低、环境污染少及可再生能源利用合理等优点,引起了各国科学家的关注。本文主要概述了电化学氮气还原(NRR)研究进展,着重讨论催化剂设计方法及原则。首先介绍NRR反应的原理和挑战,然后综述了催化剂的设计方法,通过杂原子掺杂、边缘缺陷工程、单原子活性位设计、形貌调控等方式精确设计催化剂,提高NRR反应的性能和反应效率。
1 电催化氮还原反应的原理和挑战
1.1 电催化NRR 的热力学限制
氮气分子是由2个氮原子组成的线性非极性分子(图1a)),每个氮原子的2s轨道包含一对自旋方向相反的孤电子,2p轨道含有3个自旋方向相同的孤电子(图1b))。通过原子轨道杂化,形成成键σ轨道和π轨道,反键σ*轨道和π*轨道,共用电子对在π和2σ轨道上形成高能量密度的N≡N(图1b))。N≡N稳定性高,常温常压条件下裂解具有以下挑战:1)N≡N键能高达941 kJ∙mol–1,并且第1个键的热力学裂解能为410 kJ∙mol–1,说明N≡N 解离是热力学受限过程;2)N2还原过程中,第1 个H 的加成是吸热过程(ΔH0=37.6 kJ∙mol–1),说明质子化同样是热力学受限过程;3)N2的负电子亲和能(-1.9 eV)以及高电离势(15.85 eV)提高了分子活化难度;4)最高占有分子轨道(HOMO)与最低未占有轨道(LUMO)之间的能隙大(~10.82 eV),不利于电子转移,说明氮气还原是动力学受限过程[13-14]。
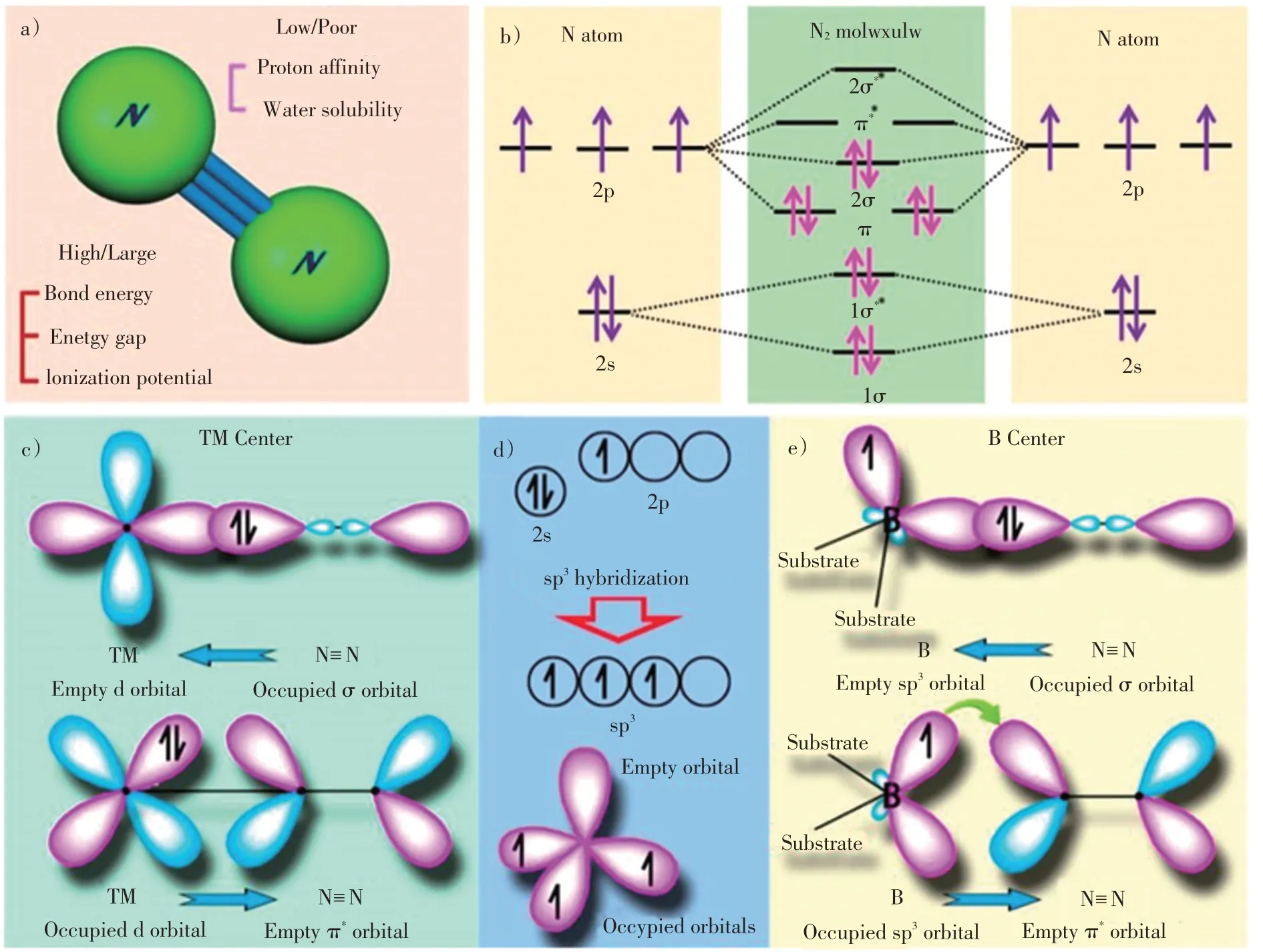
图1 a)N2的分子结构;b)N 原子轨道和N2分子轨道的示意图;c)N2与TM 结合的简化示意图;d)B 原子和具有sp3杂交的B 原子的电子构型;e)N2与B 原子成键示意图[15]Fig.1 a)Molecular structure of N2;b)schematics of N atomic orbitals and N2 molecular orbitals;c)simplified schematic of N2 bonding to TMs;d)electronic configuration of pure B atom and B atom with sp3 hybridization;e)N2 binding motifs to the B atom on the substrate[15]
1.2 氮气分子的化学吸附与活化
根据以上分析,N2的直接活化异常困难,因此,电化学NRR过程需要采用催化剂弱化氮气分子键能、降低反应过程中的活化能,从而实现低能耗、高效的固定氮气合成氨。在NRR过程中,N2分子首先吸附在催化活性中心并被活化,随后从催化活性中心接收电子,并经过一系列质子耦合过程,最后形成NH3。理论上讲,可以通过过渡金属(TMs)空d轨道与N2双电子填充的d轨道相互杂化,形成M-N键,使N2吸附在催化金属表面[2,16-17]。TMs的空d轨道接收来自氮气孤对电子后,电子对分离并转移到反键轨道,该过程被称为π back-donation机理(图1c))[15]。对于主族金属而言,空轨道和占有轨道的相互作用不能形成对称的σ 和π键,因此不能以π back-donation机理催化活化N2[18]。而非金属B基催化剂与氮气的相互作用,遵循类似的π back-donation 路线:B原子空sp2轨道接收到氮气σ轨道电子,而被占据的p轨道将电子回馈到氮气的未占据的π*轨道[19]。此外,具有sp3杂化的B原子包含占有轨道和空轨道(图1 d)),如果通过2个B-N键形式将B原子修饰到石墨相氮化碳(g-C3N4)表面,1个sp3占有轨道和1个空轨道会与N2分子形成强相互作用(图1e)),因此该类材料具有N2还原潜力[19-22]。
1.3 电催化NRR 机理
通常认为有2种电催化NRR路径:解离路径和缔合路径(图2)[14]。在解离路径(路径i)中,N≡N首先被破坏,单N原子在催化剂表面通过质子化转化为氨气。Haber-Bosch工业反应通常遵循该反应路径。根据不同的氢化序列,缔合路径(ii)可分为交替途径和远端路径。对于交替路径,2个氮原子同时发生质子化,这可能导致副产物如肼的形成。在远端路径中,末端氮原子优先参与质子化并释放氨气,表面氮原子再进行质子化。该催化路径还原产物仅为NH3,不会生成其他副产物。除此之外,还有另外1 种缔合途径,即酶路径(路径iii),在反应过程中,两个N原子同时吸附在催化剂的表面,然后依次形成NH3完成脱附[14]。除此之外,针对过渡金属氮化物(TMNs,如VN、ZrN、NbN),提出了一种遵循Marsvan Krevelen(MvK)机制的新兴催化路径:首先表面N原子被还原成NH3形成N空位,这些N空位随后被溶解的N2重新填充,随着表面N原子的还原,引发生成第2个NH3分子[23-25]。相对于解离路径和缔合路径,MvK机制需要的过电位相对较小,有利于NH3生成。

图2 NRR 的解离和缔合途径(包括远端,交替和酶促途径)的示意图[14]Fig.2 Schematic illustrations of the dissociative and associative pathways(including distal,alternating,and enzymatic pathways)for NRR[14]
2 NRR 催化剂设计
2.1 异质原子掺杂
异质原子掺杂可以调控电子结构、改变表面特性、修饰催化剂化学组成,进而调节反应底物和中间体与催化材料表面的吸附力,提高催化还原效率[26]。通常,有两类异质原子掺杂:非金属掺杂和金属掺杂。对于非金属掺杂,异质原子与毗邻的碳原子电负性的差异造成碳原子向异质原子一定程度的电子转移,导致碳基质的电荷重新分布。例如,N掺杂的碳是典型的n型半导体材料,这是因为氮掺杂后,在碳纳米管的导带引入新的电子能态,其中包括接近导带最小值的电子供态;而B在碳纳米管晶格的掺杂会在价带的最大值附近产生电子接受态,从而形成p型掺杂材料。此外,DFT计算表明,其带隙打开程度常用掺杂量呈正相关关系,因此,可通过改变掺杂原子种类和浓度调节碳基催化剂的电子态,进而产生非电中性活性位点。在诸多异质原子中,与C原子序数相近的非金属原子(如N、B、P、O等)常被优先考虑,因为相近的原子尺寸有利于异质原子与C 原子形成强化学作用,进而提高催化稳定性。通过对碳材料等的掺杂,引入自旋态和非电中性位点,进而增强对底物和中间体吸附,促进NRR[27-29]。对于金属掺杂,多是将Ru、Fe等杂原子掺杂到SnO2、TiO2等氧化物体相,均匀分散的掺杂金属原子含不饱和配位点和非对称性电荷分布,有利于底物和中间体的吸附和进一步活化,因此表现出优异的NRR活性[30-31]。
2.1.1 非金属掺杂
完整结构的碳材料如石墨呈现化学惰性,通过化学方法向其表面或体相引入氮、硼、磷等杂原子后,可以大幅提升纳米碳材料的表面化学活性。由于表面电荷状态的改变、中间体的吸附自由能的变化以及带隙的减小,碳缺陷位点可以直接充当活性位点。因此,可以在碳骨架内构建丰富的缺陷位点,使其具有良好的电化学反应性,并成为未来清洁能源的最重要选择之。
大比表面积和丰富的孔结构,可以为催化等电化学过程提供充足的结合位点和传质通道,因此N掺杂多孔碳(NPC)被广泛用于超级电容器、催化O2还原和CO2吸附等领域[32-34]。N掺杂能调节碳材料电子结构并引入缺陷和产生电荷极化,从而促进对底物的吸附和活化,有利于提高氧气还原(ORR)、NRR等催化活性[35-36]。Liu等[37]通过高温碳化以Zn为金属节点,以2-甲基咪唑为有机络合配体的ZIF-8,得到孔结构均匀分布的NPC(图3a))。研究表明,该NPC材料中N含量尤其是吡啶N和吡咯N对电催化NRR性能具有明显相关性。由750 ℃条件下煅烧得到的NPC的N含量最高,为13.5%(物质的量之比),该材料表现出最优的催化活性,-0.9 V条件下NRR法拉第效率为1.42%,NH3生成速率为1.4 mmol∙h-1∙g-1[37]。

图3 a)N 掺杂多孔碳(NPC)合成过程[37];b)B 掺杂石墨烯(BG)的透射电镜[38];c)不同BG 材料的N2脱附曲线[38];d)不同BG 材料的含B 结构比例[38];e)不同BG 材料的NRR 法拉第效率[38]Fig.3 a)Schematic illustration of NPC synthesis[37];b)TEM image of BG[38];c)N2 temperature-programmed desorption curves of graphene(G)and three BG samples[38];d)percentages of various B types in the BG samples[38];e)faradic efficiency of various samples[38]
除了N掺杂,B是另一种常用的掺杂杂原子。B(2.04)的电负性比C(2.55)的小,B掺杂会引起碳环结构电子密度重新分布,从而大大提高电催化ORR、CO2还原活性[39-40]。B原子电子云密度低,有利于吸附N2以及形成B–N键,进而发生多步质子耦合电子转移生成NH3。因此,B原子是B掺杂材料的NRR催化活性中心。另外,带正电的B 位点不容易与H+结合,有效抑制HER 竞争反应,进而提高NRR 法拉第效率。Zheng 等人合成了B 掺杂的石墨烯(BG,图3b))并研究其催化NRR 活性[38]。研究发现BG 中含有B4C、BC3、BC2O、BCO2等结构,其中BC3结构有利于N2吸附和活化(图3c)),是主要的NRR活性位点。合成的一系列样品中,BG-1的BC3含量最高(图3d)),因此具有最高的NRR催化活性(图3e)),法拉第效率高达10.8%,NH3生成速率为9.8 μg∙h-1∙cm-2。除熟知的N、B掺杂外,最近研究表明S原子掺杂同样可以增强碳纳米球(S-CNS)的NRR性能[41]。与N和B原子类似,S原子的掺杂可以改善材料对N2的物理和化学吸附能力,进而提高NRR效率。S-CNS最高NH3生成速率为19.07 μg∙h-1∙mg-1,法拉第效率为7.47%。
非金属掺杂材料催化活性受限制于低杂原子掺杂浓度,共掺杂策略可以引入新的非电中性位点,增加活性位点密度,有利于进一步提高材料电催化性能[42-46]。各种共掺杂策略如N/B,N/S 和N/P 等已被广泛用于设计有效的ORR,HER和NRR催化剂[47-49]。例如,Chen等[50]利用N/S共掺杂策略合成了N/S双掺杂碳材料(N/S-C)。N/S-C 催化NRR 速控步骤(*N2→*N2H)自由能垒为1.72 eV,比B(2.24 eV)和N(1.99 eV)掺杂材料的低。因此,该双掺杂材料具有优异的电催化NRR 活性,法拉第效率为13.79%,NH3生成速率为7.75 μg∙h-1∙mg-1。
2.1.2 金属掺杂
作为生物固氮酶活性位点,FeMo辅酶可催化N2还原成NH3[51-52]。受此启发,金属Mo掺杂引起了研究者对开发高效NRR催化剂的浓厚兴趣[53-54]。例如,Xiong等[54]合成了Mo掺杂的W18O49(Mo-WO)[54]。Mo掺杂为N2吸附提供活性位,与未掺杂材料相比,2个N原子在Mo-W中心更容易极化,电荷差显著增加(0.58 eV)。Mo掺杂引起N2吸附能由-1.65 eV增加到了-2.48 eV(图4a)和4b)),促进了电子从金属位点传递到吸附的N2,有利于进一步活化。此外,掺杂的Mo 提高了缺陷带中心费米能级,进而积累了更多的活性电子。这一系列协同效应促进了N2在Mo-WO上的吸附和活化,实现了195.5 μmol∙h–1∙g–1的高NH3产率。

图4 N2吸附在富含缺陷的a)W18O49和b)Mo-doped W18O49表面理论模型;c)在基平面和边缘平面上NRR 还原中间体结构和过渡态能量示意图;d)MoS2边缘位点NRR 能量变化示意图;e)MoS2催化NRR 稳定性测试[55]Fig.4 The optimized adsorption configurations of N2 molecules with their corresponding charge distribution on the surface of a)defect-rich W18O49 and b)Mo-doped W18O49 models;c)schematic structures and energies of the intermediates and transition states for NRR on the basal and edge planes;d)freeenergy profile for NRR at MoS2 edge site;e)stability evaluation with 10 successive tests[55]
在生物固氮酶中,Fe是另一种重要金属元素,也常被用作掺杂剂以提高材料催化性能。例如Sun课题组[56]将Fe掺杂到TiO2得到高效的NRR催化剂。Fe的引入增加了TiO2中O空位含量。该O空位可以向Ti4+传递电子形成活性的Ti3+,进而提高材料NRR催化效率。因此,Fe掺杂的TiO2表现出优异的NRR性能,法拉第效率高达25.6%,NH3生成速率为25.47 μg∙h-1∙mg-1。
由于Zr4+的d电子构型与TiO2的类似,Zheng 课题组[57]将Zr4+掺杂到TiO2中设计新型NRR 电催化剂(Zr-TiO2)。与利用低价金属掺杂形成O空位不同,稳定的Zr4+掺杂引入了拉伸应变和压缩应变,从而促进了大量与bi-Ti3+位点相邻的O空位形成。正如理论计算所揭示的,Zr-TiO2(101)面的这些bi-Ti3+位点可以有效增强N2的化学吸附。Zr-TiO2的NRR法拉第效率为17.3%,NH3生成速率为8.90 μg∙h-1∙cm-2。
2.2 边缘工程(Edge Engineering)
边缘工程在调节诸如石墨烯、氮化物或碳氮化物(MXenes)等材料的生长动力学和形态调控中起着至关重要的作用[58-59]。边缘结构调控可赋予材料不同的电子结构和功能,创造更多的活性位点,是设计理想NRR催化剂的有效手段之一[60-62]。
MXenes作为一种新型材料已广泛用于分离,能量存储与转化等领域[63-65]。理论研究表明,终端结构由M原子组成的MXenes(如V3C2和Nb3C2)材料能够活化N2分子,在电催化NH3合成方面具有巨大的潜力[66]。然而,当材料终端与含氧基团(如OH*和O*)相连时,MXene 对N2的结合能力弱,导致NRR 催化效率低,主要发生HER反应[67]。而在边缘平面暴露足够多活性位点的结构,例如Ti3C2Tx(T=O,F)MXene的T-Ti-C-Ti-C-Ti-T结构,具有高效NRR 催化活性。DTF 理论研究表明,边缘平面的中间Ti 原子能以较低活化能(*N2→*NNH过程活化能为0.64 eV,*NNH →*NNH2为0.52 eV)催化NH3形成(图4c))。
类似地,MoS2的边缘结构被证明是极化和活化N2分子的关键部位[55,68]。带正电的Mo-edge促进了Mo-N键的形成,并使N/N键的长度从*N2中的1.129 Å 延长到*NNH中的1.221 Å。理论计算研究表明,速控步骤(*NN →*NNH)能垒仅为0.68 eV(图4c))。实验结果研究表明,MoS2具有催化NRR活性,其法拉第效率为1.17%,NH3产率为80.8 pmol∙s–1∙cm–2,并且连续10次循环测试法拉第效率无明显变化(图4d))。
2.3 结构调控
2.3.1 单原子催化剂
鉴于催化剂尺寸对活性的影响,文献报道了众多催化NRR的金属纳米粒子、纳米簇、纳米晶和金属氧化物量子点等[69-70]。当前,大多数电催化体系仍依赖贵金属基催化剂(Ru、Au等),而催化反应仅在有限活性位点的外表面发生[71-72]。因此,提高金属利用效率、减少金属消耗,对降低催化剂成本,促进可持续性至关重要[73]。在过去几年中,越来越多的研究集中在小尺寸催化剂制备,从纳米颗粒、亚纳米尺寸团簇到单原子催化剂(SAC)[74]。其中,SAC 因拥有最大的原子利用效率,不饱和活性位点和独特的尺寸量子效应,引起了研究者的兴趣,并成为多相催化反应中最有吸引力催化材料之一[75-79]。金属原子与碳原子之间的结合能低,因此需要在碳骨架上建立配位能力强的锚定位点捕获金属原子。最常见的锚定位点为吡啶型及石墨型N原子,它们分别与2或3个sp2杂化的C原子相连,并在π系统中分别提供1或2个电子,提高碳表面对金属原子的锚定能力,从而进一步形成金属-氮配位键,增加活性位点稳定性。金属原子均匀分散在载体上,使SAC与常规催化剂相比,催化活性得到显著提高。例如,Geng等[80]以含Ru ZIF-8为前驱体,通过高温煅烧的方法合成Ru基单原子催化剂Ru SAs/N-C。XANES 数据显示Ru原子以Ru-N 键形式均匀分散在碳基底上,价态为+3价。原子光谱分析测试表明Ru原子的负载量为0.18%。该催化剂在-0.2 V电位下,催化NRR法拉第效率高达29.6%,产NH3速率为120.9 μg∙mgcat-1∙h-1,为当时报道最高值。化学吸附测试显示碳化后的含Ru材料强N2吸附能力,有利于N2吸附和活化。N2在Ru SAs/N-C单原子催化剂Ru-N3和Ru-N4结构上的吸附能分别为0.73 eV和0.77 eV,低于在纳米金属Ru(101)的吸附能(0.91 eV),进一步解释了该单原子催化剂的高活性和选择性。类似地,以含未配位-NH2的UiO-66-NH2为基底,通过氨基对Ru3+的分散稳定作用,合成有单原子催化剂Ru@ZrO2/NC[81]。高温碳化后Ru的负载量为0.1%。该催化剂在-0.21 V电位下催化生成NH3速率为3.665 mg∙h-1∙mgRu-1。理论计算表明具有氧空位的Ru 单原子位点有利于N2吸附,能稳定*NNH 中间体,是催化活性中心。除了贵金属和稀有金属基SAC,过渡金属Fe、Cu 基SAC 在催化NRR方面也受到了极大关注[82-83]。例如,由聚吡咯Fe碳化得到的FeSA-N-C(Fe@NC)催化剂电催化NRR法拉第效率高达56.55%,NH3生成速率为7.48 μg∙h-1∙mg-1[84]。理论计算研究表明,Fe配位态在NRR催化过程中起着关键作用,高度自旋极化的FeN3活性明显高于FeN4[85]。除了Fe原子的化学配位态外,在理论研究中还应考虑其他潜在因素,例如支撑基质和电解质,从而准确估计SAC的整体性能。
2.3.2 形貌调控
除了催化剂尺寸对活性影响,另一种增强催化活性的有效策略是可控地合成具有目标形貌的催化材料,例如1D超细纳米线/纳米纤维,2D超薄纳米片和3D多孔/空心结构等[86]。其中,1D纳米结构因其独特的物理化学性质而在催化领域受到广泛关注[87]。1D纳米结构可以提供电子传递途径,进而促进电子传输。大比表面积使N2与催化电极在电解质/催化剂的界面处能够充分相互作用,从而改善界面传质。此外,原位阵列生长有利于提供更多活性位点,增强催化剂在基底上的结合强度。MacFarlane 教授[88]利用热还原β-FeOOH 的方法将α-Fe 纳米矩阵原位生长在碳纤维电极表面。该复合材料具有较小的*N2H 生成自由能(ΔG=0.15 eV),能在常温、常压条件下实现电催化合成NH3。
超薄2D纳米片具有独特物理、电化学性质[89]。大横向尺寸和有限层数使其具有超高的比表面积,有利于促进表面相关的催化反应。同时,高度暴露的表面原子使超薄2D纳米片易于表面修饰和功能化。如前所述,这些特点将促进N2在超薄2D纳米片催化剂上的吸附和活化。尽管在NRR催化应用潜力巨大,但2D纳米材料相关的研究才刚刚开始。
3D多孔结构可以有效缩短电子传递距离,增加底物吸附保留时间,促进催化转化。Zhang等[90]以泡沫镍作为还原剂和基底,合成了S修饰的多孔Au电极,发达的孔隙结构是底物传质通道,并且电子不需要经过体相传递而是在孔内与底物N2结合,完成催化转化。S掺杂有利于N2选择性质子化生成NH3。中性条件下,该电解催化N2还原的法拉第效率为17.2%,NH3生成速率高达22.7 μg h-1mg-1cat.。
3 总结与展望
电催化NRR以可再生电能为驱动力,将含量丰富的N2和H2O转化为高附加值化学品氨气,不仅是替代传统Haber-Bosch工艺的理想选择,还是以化学键的形式储存可再生能源的有效方式之一。自2015年,科学家通过对催化剂设计、电解液及电解池设计与探索,提高NRR 活性及选择性。本文对电化学氮气还原(NRR)机理和热力学限制进行讨论,并对高效催化剂开发策略做了详细综述。目前该研究领域已经取得了一定研究进展,但仍面临着巨大挑战。
规范实验操作,排除外源污染。就测试技术而言,N2在电解液溶解度低,生成NH3的总浓度与环境污染浓度在1个数量级,使定量分析NH3生成效率具有挑战,阻碍了该研究领域的快速发展。NRR的污染源主要来自测试系统内和系统外的污染。系统内的污染通常会对氨产量产生显著影响,甚至产生假阳性结果。系统内的污染主要有原料气、电解液、催化剂以及Nafion膜等中的含氮化合物。为了排除假阳性实验结果,需要做以下对照实验。排除电解液、催化剂氨污染:必须在与NRR实验完全相同的条件下用氩气来代替氮气,以便量化来自电解液或催化剂本身污染的氨。排除原料气氨污染:在开路电位测试条件下,与NRR实验保持相同的电解时间,研究原料气中污染氨量。排除NOx-影响:用于制备电解液的锂盐中含有一定量的NOx-。Li2SO4的沸点为1377 ℃,远高于LiNO3(600 ℃)和LiNO2(350 ℃)的沸点,因此可以在氩气环境中对Li2SO4进行800 ℃高温退火处理,来消除Li2SO4中的硝酸盐或亚硝酸盐,并进一步验证氨的来源。排除测试体系氨污染:离子交换过程中,Nafion膜会积累一定量的铵离子,每次使用之前需要用大量的高纯水冲洗。系统外的污染主要包括空气中的氨或硝酸盐、测试者的呼吸及橡胶手套。为了消除系统外污染,所有电化学装置和消耗品都应该在NRR实验之前彻底清洗。总之这些系统外的污染必须通过使用完全封闭的系统,严格控制实验条件来合理的排除。同位素标记验证N源:在经过系统研究寻找到最佳实验条件后,为了更确切地证明检测到的氨来源于氮气电还原过程,需要使用同位素标记的15N2气体进行实验,然后定量检测产生15NH3。在成功观察到NRR的实验现象后,需要重复实验,确保数据可信度在误差允许范围内。
耦合材料设计策略,开发高效NRR催化剂。未来催化剂设计需要依赖多种设计策略,例如对杂原子掺杂材料进一步进行形貌调控,以暴露更多的活性位点,促进电子迁移,并增强反应物和中间体与催化剂结合力。开发先进的表征手段,特别是原位表征手段,对于监测反应中间体的转化和催化剂表面结构的变化至关重要。理论计算结合实验数据,可以对反应过程和结构与性能之间的关系有更深入的了解,有助于高效催化剂的精确设计。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
生物学通报(2019年3期)2019-02-17
电脑知识与技术(2018年19期)2018-11-01
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
