
脂肪细胞因子对代谢的调节
2022-09-06 13:38周伟玉汤其群
中国生物化学与分子生物学报 2022年6期
周伟玉, 汤其群, 刘 洋
(复旦大学基础医学院代谢分子医学教育部重点实验室, 上海 200032)
脂肪组织通常认为是能量储存库,但在新的研究中发现其也是内分泌组织[1]。脂肪细胞因子(adipokine)是脂肪组织产生并分泌的具有一定生物活性的物质,绝大部分为肽类物质。目前,已有的研究发现,脂肪组织是600多种潜在分泌蛋白质的来源[2]。脂肪细胞因子包括瘦素(leptin)、脂联素(adiponectin)、抵抗素(resistin)、骨形成蛋白4(bone morphogenetic protein 4,BMP4)、锌-α2-糖蛋白(zinc-α2-glycoprotein,ZAG)、成纤维细胞生长因子21(fibroblast growth factor 21,FGF21)、内酯素(visfatin)等,具有调节脂肪分布、胰岛素分泌和敏感性、能量消耗等功能[3]。脂肪细胞因子可对机体多种代谢,特别是糖脂代谢具有一定的调控功能。本综述将简要介绍部分脂肪细胞因子对代谢的影响。
1 瘦素对代谢的影响
1.1 瘦素
1994年瘦素的发现改变了人们对脂肪组织的看法,使人们认识到脂肪组织不只是能量储存库,还是一种内分泌组织[4]。瘦素主要来源于脂肪组织,被认为是能量稳态的基本调节器,调节外周组织中的葡萄糖代谢和脂质代谢[5]。瘦素受体(leptin receptor,LepR)在下丘脑和下丘脑外的几个脑区中有表达。瘦素通过瘦素受体的长信号形式发挥作用,而瘦素受体基因编码的各种截断类型的受体,不具有完全信号转导能力。瘦素和其受体结合后,Janus激酶2(Janus kinases 2,JAK2)被激活,从而磷酸化瘦素受体上3个不同的酪氨酸残基(Tyr985、Tyr1077和Tyr1138)。1 138位点酪氨酸残基的磷酸化能促进信号转导子和转录激活子3(signal transducer and activator of transcription 3,STAT3)的募集,随后被JAK2磷酸化。磷酸化的STAT3转移到细胞核并激活靶基因转录。血浆中的瘦素水平与能量储存有关,因此肥胖时瘦素增加,禁食时减少[4]。瘦素或者瘦素功能性受体的缺失(Lepob/ob或LepRdb/db小鼠)与高血糖、高胰岛素血症和葡萄糖耐受显著相关。通过对以往瘦素调查数据重新统计分析发现,体内瘦素浓度是分位数依赖的。这一发现解释了相对于男性,基因对女性瘦素浓度有更大的影响[6]。
瘦素对神经支配的影响,是通过下丘脑弓状核刺鼠相关蛋白(agouti-related peptide,AGRP)神经元和阿黑皮素原神经元介导的。在这两类神经元中,编码LepR基因的缺失会导致脂肪组织中神经分布的减少[7]。并且AGRP神经元上γ氨基丁酸的传入是瘦素急性抑制食欲所必需的[8]。在正常情况下,外周分泌的瘦素会通过血脑屏障被运输到脑中目标部位,特别是下丘脑。然而,肥胖会导致瘦素在血脑屏障中的渗透减少,鼻内联合给药两性细胞穿透肽,可以有效地将瘦素传递到大脑,用于肥胖的预防和治疗[9]。高脂饮食诱导的肥胖小鼠下丘脑中蛋白酪氨酸磷酸酶1B(protein tyrosine phosphatase 1B,PTP1B)和T细胞蛋白酪氨酸磷酸酶(T cell protein tyrosine phosphatase,TCPTP)含量增加,而瘦素含量和胰岛素敏感性降低。小鼠鼻内给药靶向抑制PTP1B和TCPTP会增加瘦素含量和胰岛素敏感性,并通过抑制进食和增加能量消耗降低体重[10]。尽管瘦素在改善代谢方面具有强大功效,但是临床上的瘦素疗法未能有效治疗肥胖,主要原因是出现瘦素抵抗现象。瘦素抵抗是一个复杂的现象,涉及到脑脊髓室对瘦素摄取的缺陷,以及中枢神经元中瘦素信号的衰减[11]。而部分降低肥胖小鼠血浆中瘦素的水平可以恢复下丘脑瘦素的敏感性,并且有效地降低体重增加和提高胰岛素敏感性[12]。
1.2 瘦素对血糖的调节作用
瘦素通过中枢神经系统发挥抗糖尿病的作用,包括增加外周组织葡萄糖的摄取和减少肝和肾葡萄糖输出[13]。但是瘦素如何通过神经系统将效应传递到外周组织,导致葡萄糖摄取和氧化增加,目前尚不清楚。有人认为,1型糖尿病的一些代谢表现可能是由于瘦素含量降低而引起的。在1型糖尿病小鼠模型中,给予瘦素能够预防高血糖,这表明对瘦素分泌的适当控制对于正常生理活动至关重要[14]。低剂量的瘦素并未减轻体重,却能有效地使血糖和胰岛素浓度正常化,这表明瘦素在调节血糖水平方面比抑制食欲更有效。Lepob/ob小鼠在体重未发生任何变化前,瘦素可以在15分钟内迅速改善体内葡萄糖的平衡。表明瘦素可以有效地改善葡萄糖稳态,并且不需要瘦素诱导体重下降和体脂减少。瘦素通过激活胰岛素受体1使胰岛素信号变得敏感,导致该分子对胰岛素信号转导的亲和力更强,从而调节了血糖水平。当小鼠摄入过多盐分时,肝和下丘脑的醛糖还原酶-果糖激酶通路会被激活,导致内源性果糖含量增加,发生瘦素抵抗[15]。
1.3 瘦素对脂质的调节作用
在心脏中,瘦素通过优先增加甘油三酯(triglycerides,TG)脂解和游离脂肪酸(free fatty acids,FFA)氧化改善代谢[16]。而瘦素在肌肉中会发生胰岛素抵抗影响肌肉对FFA的利用[17]。在Lepob/ob和Lepob/+小鼠中,高密度脂蛋白(high-density lipoprotein,HDL)在白天达到高峰,而在LepRdb/db和LepRdb/+小鼠中,血浆HDL在夜间达到高峰。小肠和肝来源的大部分脂质通过合成和分泌含有脂蛋白的载脂蛋白B(apolipoprotein B,apoB)运输到外周组织中。这些脂蛋白质的合成需要一种细胞内伴侣,即微粒体甘油三酯转移蛋白(microsomal triglyceride transfer protein,MTP),它可以调节脂肪在不同组织中的动员。研究表明,瘦素与小肠内高表达的长型LepR相互作用,下调MTP的表达;然而,在肝中,瘦素与短型LepR相互作用,上调MTP的表达[18]。瘦素通过血脑屏障到达大脑,利用迷走神经背侧复合体的信号传导,增加肝内TG的分泌,并减少肝中脂肪的从头生成。这些中枢介导的瘦素效应需要完整的肝迷走神经支配,在高脂饮食条件下,向大鼠大脑注射瘦素,瘦素对肝内良好代谢表型的作用仍然存在[19]。
2 脂联素对代谢的影响
2.1 脂联素
脂联素是含量最丰富的分泌型脂肪细胞因子,自1995年首次被发现以来,就受到科学界的极大关注[20]。脂联素是一个30 kD的单分子糖蛋白,通过靶细胞上的脂联素受体1(adiponectin receptor 1,AdipoR1)和脂联素受体2(adiponectin receptor 2,AdipoR2)发出信号[20]。女性脂联素的循环水平比男性高约40%,原因可能是雄激素对脂联素有抑制作用。血清中脂联素水平随着肥胖程度的增加而降低,并与胰岛素敏感性呈正相关[21]。高脂饮食后机体内FFA含量升高,中枢神经系统的巨噬细胞——小胶质细胞会引发下丘脑炎症。而脂联素通过调控小胶质细胞活性抑制FFA引发的下丘脑炎症,维持机体能量稳态[22]。脂联素可以通过活化一磷酸腺苷活化蛋白激酶(AMP-activated protein kinase,AMPK)和p38α阻止肝癌的发展,小鼠和人类的睾酮可以激活脂肪细胞中的c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)以抑制脂联素分泌,增加肝癌细胞的增殖[23]。
肥胖个体体内循环脂联素浓度会降低,这种现象被认为在与肥胖、动脉粥样硬化和心血管疾病的发病机制中具有关键作用。而体重减轻可以显著提高血浆中脂联素的水平[24]。尽管脂联素基因主要在脂肪细胞中表达,但最近的研究发现,脂联素基因也可以在肝细胞[25]和骨骼肌[26]中被诱导表达。
2.2 脂联素对血糖和脂质代谢的调节作用
人体内大约90%的循环脂联素以两种形式存在:一种是由12或18个脂联素分子组成的高分子量(high molecular weight,HMW)结构(360~540 kD),另一种是由6个脂联素分子组成的低分子量(low molecular weight,LMW)结构(180 kD)。人体内剩余10%的脂联素,以脂联素三聚体的形式存在,被称为全长脂联素三聚体(90 kD)。腹腔注射HMW和LMW脂联素可以降低健康小鼠、1型糖尿病和2型糖尿病小鼠的血糖含量。脂联素通过降低肝内葡萄糖产生的限速酶来抑制糖异生[27]。除此之外,脂联素还能通过减少对糖异生底物的利用来抑制葡萄糖的产生[28]。脂联素含量的减少会导致1型糖尿病和2型糖尿病患者体内葡萄糖异常增加[29]。
脂联素通过增加内脏白色脂肪组织中的脂蛋白脂肪酶活性,导致内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)/AMPK激活和肌肉中的脂质氧化,降低肝和肌肉中的脂质异位堆积,从而逆转高脂饮食喂养引发的小鼠胰岛素抵抗。这些作用会降低质膜二酰基甘油含量,从而降低肝中蛋白激酶Cε(protein kinase Cε,PKCε)的活化和肌肉中PKCε/PKCθ的活性,从而改善这些组织中的胰岛素信号[30]。
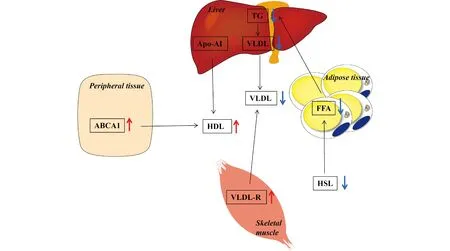
Fig.1 Regulation of adiponectin on lipid metabolism[31] Adiponectin increases the content of HDL by up-regulating the expression of ABCA1 in peripheral tissues and the production Apo-AI in the liver. Adiponectin also indirectly reduces release of FFA from the adipose tissue to the liver by inhibiting lipolysis in adipocytes. This effect results in the reduction of VLDL production due to the decreased of TG in the liver. Adiponectin-induced up-regulation of VLDL-R in skeletal muscles also leads to the reduction of VLDL in blood. HDL,high-density lipoprotein;ABCA1,ATP-binding cassette transporter A1;Apo-AI,apolipoprotein-AI;FFA,free fatty acids;VLDL,very low density lipoprotein;TG,triglyceride;VLDL-R,very low density lipoprotein-receptor;HSL,hormone-sensitive lipase
脂联素通过上调外周组织中ATP结合盒转运蛋白A1(ATP-binding cassette transporter A1,ABCA1)和肝中载脂蛋白AI(apolipoprotein-AI,Apo-AI)的表达,增加HDL的含量[31]。脂联素也通过抑制脂肪细胞的脂解,间接使脂肪组织向肝内运输的FFA含量下降,导致肝中TG含量的降低,进而减少极低密度脂蛋白(very low density lipoprotein,VLDL)的含量。脂联素诱导的骨骼肌中极低密度脂蛋白受体(very low density lipoprotein receptor,VLDL-R)含量的增加,也会使血液中VLDL含量降低[31](Fig.1)。
2.3 脂联素对胰岛素敏感性的调节作用
脂联素通过对蛋白磷酸化激活因子受体-α的激活增加FFA的代谢和能量消耗,导致肝和骨骼肌中TG含量降低,从而增加胰岛素敏感性[32]。神经酰胺是参与胰岛素抵抗、细胞死亡、炎症和动脉粥样硬化等多种生理活动的脂质[33]。脂联素的使用会导致肝中神经酰胺水平的显著降低,从而增加胰岛素含量。血浆中脂联素水平的降低可能与脂肪代谢相关的胰岛素抵抗有一定的关联[34]。脂联素促进葡萄糖刺激的胰岛素分泌,防止胰腺细胞的细胞凋亡并促进胰腺细胞的生存。流行病学数据显示,脂联素是预测个体代谢中胰岛素敏感性最强的细胞因子[35]。
脂肪代谢障碍和胰岛素抵抗的小鼠会出现高胰岛素血症、高血糖症,并且它们的血清中无脂联素。系统破坏AdipoR1会导致小鼠葡萄糖产量的增加,然而破坏AdipoR2小鼠葡萄糖摄取减少,胰岛素抵抗增加[36]。对AdipoR1和AdipoR2双敲除的小鼠注射脂联素,并不会导致小鼠血糖降低。低蛋白质饮食会提高大鼠的胰岛素敏感性,增加体内循环脂联素的总含量。肝和骨骼肌是脂联素对胰岛素敏感作用的主要靶点。在分离的大鼠原代肝细胞中,脂联素具有增强胰岛素抑制葡萄糖生成的能力[37]。脂联素增加肝中胰岛素的作用,部分是通过刺激AMPK的磷酸化,导致乙酰辅酶A羧化酶(acetyl CoA carboxylase,ACC)磷酸化的增加,以及磷酸烯醇式丙酮酸羧化激酶(phosphoenolpyruvate carboxykinase,PEPCK)和葡萄糖-6-磷酸酶(glucose 6 phosphatase,G-6-Pase)的活性降低而发挥作用。对小鼠腹腔注射脂联素会降低全身葡萄糖和FFA浓度,而不改变胰岛素的浓度[37]。在孕妇中,脂联素可以增强骨骼肌的胰岛素敏感性和葡萄糖的摄取,降低胎盘转移所需的营养供应[37]。
脂联素优先促进脂肪储存在皮下脂肪垫,而不是内脏脂肪垫,肝或者骨骼肌中[38]。这种优先的脂肪储存方式减少了内脏脂肪组织的堆积和全身炎症,改善了葡萄糖和脂肪代谢,增加了胰岛素敏感性。促炎脂肪细胞因子/细胞因子也能显著影响胰岛素信号,特别是当它们导致脂肪组织炎症时。脂肪细胞和基质血管细胞中脂联素降低了肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)和白介素-6的释放[39]。相反,脂联素刺激抗炎脂肪细胞因子白介素-10的释放[40]。肥胖与脂肪细胞炎症、巨噬细胞浸润以及巨噬细胞极性由M2表型(抗炎)向M1表型(促炎)转变有关[41]。研究发现,脂联素可以将巨噬细胞的极性转移至M2表型[42]。除了调节巨噬细胞的极性转化,脂联素还抑制髓系祖细胞的生长,抑制巨噬细胞的功能,包括吞噬作用和脂多糖诱导的TNF-α的产生[43]。
3 抵抗素对代谢的影响
3.1 抵抗素
抵抗素是Steppan等[44]于2001年提出,其是对胰岛素具有抗性的脂肪细胞因子,被认为是2型糖尿病死亡的预后因素[45]。影响生物体内细胞因子和脂肪细胞因子水平的因素之一,是它们对应基因的启动子和内含子区域的功能多态性。体内抵抗素70%左右的变化是由遗传因素和单核苷酸多态性所引起的。啮齿动物的抵抗素主要由脂肪细胞产生[46],而人类的抵抗素主要由外周血单核细胞产生[47]。抵抗素最初被认为是通过降低小鼠胰岛素敏感性和葡萄糖耐受量来发挥作用。而最近关于啮齿动物的研究结果表明,抵抗素主要是通过作用于肝而发生胰岛素抵抗[48]。
3.2 抵抗素对胰岛素抵抗和脂质代谢的调节作用
啮齿动物在高脂肪/高碳水化合物喂养的肥胖状态下,体内循环抵抗素水平升高,在生理上表现为葡萄糖摄取受损和胰岛素敏感性下降。抵抗素通过AMPK依赖性和AMPK非依赖性细胞因子信号转导抑制因子3(suppressor of cytokine signaling-3,SOCS-3)的信号通路促进胰岛素抵抗。Palanivel等人的研究表明,抵抗素可提高FFA的稳定性诱导肝发生胰岛素抵抗[49]。
在正常情况下,胰岛素会抑制肝内葡萄糖的产生,而抵抗素已经被证明可以通过促进血糖水平的升高降低胰岛素的作用。而运动会下调肺腺癌转移相关转录本1(metastasis-associated lung adenocarcinoma transcript 1,MALAT1)的含量,并通过升高microRNA-382-3p和降低抵抗素的含量,缓解2型糖尿病患者的人脐静脉内皮细胞的胰岛素抵抗[50]。增加肝内糖异生率可能是2型糖尿病患者抵抗素发挥作用的关键步骤。用抗抵抗素的抗体IgG中和抵抗素能提高饮食诱导的肥胖小鼠的胰岛素敏感性[44]。禁食后,抵抗素敲除的小鼠表现出葡萄糖水平和肝内糖异生酶表达降低[51]。Brown等[52]研究了抵抗素对胰岛素的分泌、胰岛素受体基因的表达、胰岛β细胞的功能和体外生存能力的影响。他们的结果显示,抵抗素导致胰岛素受体在mRNA和蛋白质水平表达显著降低,但不影响胰岛素的分泌。二甲双胍可以改善糖尿病所引起的高血糖、高胰岛素血症等不良反应,但是也发现在二甲双胍治疗后,抵抗素在蛋白质水平上有所增加。
抵抗素在小鼠3T3-L1细胞和原代前脂肪细胞分化成熟过程中被诱导表达,外源重组小鼠抵抗素可以促进3T3-L1细胞的脂质生成,而敲低抵抗素可显著降低3T3-L1脂肪细胞分化后的脂质含量。褪黑素通过对脂肪细胞中大脑和肌肉ARNT样蛋白-1(brain and muscle arnt-like protein 1,Bmal1)的转录抑制和N6-腺苷酸甲基化RNA的去甲基化,减少脂肪细胞来源的外泌体中抵抗素的含量,并通过AMPKα信号缓解内质网应激诱导的肝脂肪变性[53]。
4 骨形成蛋白4对代谢的影响
4.1 BMP4
BMP4是转化生长因子β超家族的一员,定位于细胞外,以自分泌和旁分泌的方式发挥作用[54-56]。RNA测序显示,BMP4是糖尿病视网膜病变的治疗靶点[57]。虽然BMP4最初是由其促进成骨能力而被发现,但现在认为其对胚胎的生长发育和成年人组织稳态也具有关键作用。人类皮下脂肪组织前脂肪细胞在分化过程中会诱导BMP4的产生,并且BMP4可以增加前脂肪细胞定向和分化。在人类前体细胞的研究中发现,BMP4也可以诱导米色表型的出现。肥胖患者和小鼠,其BMP4的水平均有明显增加,但BMP4水平的增加是肥胖的原因还是结果仍有待于确定。BMP4可能通过抑制哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)信号通路抑制肝的脂肪变性,减缓非酒精性脂肪肝的进程[58]。BMP4通过促进糖酵解来保护肝癌细胞免受缺氧和低血糖的影响,因为BMP4上调了葡萄糖摄取、乳酸生成、ATP水平和糖酵解限速酶(包括己糖激酶2和磷酸果糖激酶)的活性[59]。对已经发生肥胖的小鼠进行BMP4基因治疗。结果显示,虽然BMP4并未减轻肥胖小鼠体重,但改善了全身胰岛素敏感性,增强了所有关键代谢组织的胰岛素信号,并降低了肝中关键的糖异生酶的表达[60]。
4.2 BMP4对脂肪细胞的代谢调控
BMP4可与其他发育信号一起调控骨髓间充质干细胞(bone marrow stem cells,BMSCs)的命运[61]。BMP4在前棕色脂肪细胞中表达,在终末分化过程中表达逐渐减少。此外,与成熟脂肪细胞相比,BMP4在基质血管成分中的表达量更高,说明BMP4在棕色脂肪细胞中主要来源于干细胞微环境、BMSCs和前脂肪细胞[62],这对启动和促进脂肪生成是十分重要的。
棕色脂肪组织中,BMP4的水平明显低于白色脂肪组织[63]。BMP4对棕色脂肪组织和白色脂肪组织的作用不同,但总体呈现的效果是胰岛素敏感性增加。正在分化的棕色脂肪细胞可以分泌BMP4的抑制剂来应对BMP4水平的升高。同时,BMP4会使棕色脂肪组织中解偶联蛋白1(uncoupling protein 1,UCP1)含量降低,脂质含量增加。BMP4诱导皮下白色脂肪组织发生米色化,改善胰岛素敏感性,减少肝的脂肪堆积和脂肪组织炎症[63]。敲除BMP4可以增强白色脂肪组织中雌激素受体α的水平,降低成年雌鼠高脂饮食诱导的肥胖和胰岛素抵抗[64]。脂肪组织中BMP4含量的增加会导致皮下脂肪的积累和内脏脂肪质量的变小。敲除脂肪组织中的BMP4会导致成年雄性小鼠皮下和内脏脂肪细胞变大,而雌鼠中并未出现这种现象。BMP4可以增强M2型巨噬细胞诱导米色脂肪形成的能力。因此,通过BMP4调控脂肪组织巨噬细胞诱导脂肪米色化可能是治疗肥胖的一种新的策略。
5 锌-α2-糖蛋白对代谢的影响
5.1 ZAG
ZAG最先分离自人的血浆中,由各种器官例如肝、乳腺和肺,尤其是脂肪组织分泌,被认为是一种肿瘤衍生的癌症恶病质[65]和2型糖尿病患者肾病的早期生物标志物[66]。ZAG作为白色脂肪组织的脂质动员因子,其高表达会使癌症恶病质患者体重减轻[67]。此外,ZAG似乎还与胰岛素敏感性和棕色脂肪组织的产热有关[68]。糖皮质激素、β3肾上腺素能受体激动剂、甲状腺激素和生长激素等因素会增加脂肪组织中ZAG的表达。ZAG在脂肪组织中以旁分泌或自分泌的方式发挥作用,通过刺激β3肾上腺素能受体增加脂肪的分解和产热。高脂喂养条件下,ZAG基因缺失的大鼠相对于正常大鼠体重增加和脂解减少更加显著,即使使用β3肾上腺素能受体激动剂治疗后也未见恢复[69]。慢性低度炎症和血清瘦素浓度的升高会减少脂肪组织中ZAG的分泌[70-72]。
ZAG的血浆浓度受体重和健康状况多种因素影响。研究表明,女性在出生最初几个月中肥胖程度的增加与脐带血中ZAG浓度成反比。但这一结果在男性中并未观察到,表明ZAG对体重的调节与性别相关[73]。线粒体脱氧核糖核酸(mitochondrial DNA,mtDNA)调节线粒体氧化磷酸化,并且每个线粒体可以识别多个拷贝的mtDNA[74]。过表达ZAG后肝的mtDNA拷贝数增加,表明ZAG可以增加线粒体的数量并提高线粒体的功能[75]。
5.2 ZAG与胰岛素敏感性
ZAG通过刺激脂肪细胞中脂联素的分泌,从而增加外周器官胰岛素的敏感性[76]。ZAG可以通过激活AMPK,促进葡萄糖转运蛋白4的表达,增加葡萄糖的摄取[77]。过表达ZAG可减轻高脂饮食诱导小鼠胰岛素抵抗,同时降低骨骼肌脂质含量[78]。对Lepob/ob小鼠给药ZAG会直接抵抗肥胖和糖尿病[68, 79]。相比正常男性,肥胖久坐的男性其皮下脂肪中ZAG的mRNA和蛋白质水平显著降低。但是,肥胖所引发的皮下脂肪中ZAG表达下降并未影响血清中ZAG含量的改变,这一结果预示,皮下脂肪来源的ZAG对整体ZAG循环水平无显著影响。
5.3 ZAG对脂质代谢的调节及对白色脂肪细胞棕色化的调控
ZAG在非酒精性脂肪肝(non-alcoholic fatty liver disease,NAFLD)患者和小鼠的肝组织中的表达显著下调。在人肝细胞LO2细胞中,ZAG通过阻断TNF-α信号介导的炎症反应和细胞内脂质沉积,促进细胞增殖,抑制细胞凋亡以减轻NAFLD的作用[80]。在HepG2细胞中过表达ZAG显著抑制脂肪生成,促进脂肪分解和脂肪酸β氧化,并减弱棕榈酸诱导的细胞内脂质堆积。相反,下调ZAG的表达会显著抑制脂肪酸β氧化,增加脂肪生成和脂质积累[81]。肥胖大鼠过表达ZAG会诱导激素敏感性脂肪酶(hormone-sensitive lipase,HSL)的表达,减少内脏脂肪中脂肪酸合成酶(fatty-acid synthase,FAS)和ACC的表达[82]。这些结果表明,ZAG改变体重的机制涉及脂肪生成和脂肪分解相关酶的调节。
在3T3-L1细胞中,过表达ZAG可以显著上调UCP1和其他棕色脂肪细胞标记物PR结构域16(PRdomain-containing the 16-like protein,PRDM16),以及细胞死亡诱导DNA断裂因子α类似效应A(cell death induced by DNA fragmentation factor alpha A,Cidea)的表达,表明ZAG在诱导细胞棕色化中可能发挥作用[83]。在中国受试人群中,皮下白色脂肪组织和血清中ZAG的含量与身体质量指数(body mass index,BMI)和体重成负相关。ZAG对肥胖小鼠体重、脂肪重量和糖代谢的作用可能是通过激活皮下白色脂肪组织和内脏白色脂肪组织中过氧化物酶体增殖物激活受体γ辅激活子1α(peroxisome proliferator-activated receptor γ coactivator 1-alpha,PGC-1α)的表达,进而促进白色脂肪组织的棕色化而实现的[84]。
6 问题与展望
脂肪组织可以分泌数百种具有生物活性的分子,其中大多数是多肽类物质。脂肪细胞因子是参与饱腹感、能量消耗、脂肪分布、脂肪细胞功能、葡萄糖和脂质代谢、胰岛素敏感性和其他生物过程的重要调节剂(Fig.2)。近年来,脂肪细胞因子与生物机体代谢之间的新关联被不断发现。已有针对脂肪细胞因子开发的一系列治疗代谢性疾病的方案,但在这进程中充满了困难与挑战,其中重要原因是人体的复杂性,单一脂肪细胞因子含量的改变,可能会引起其他因子的代偿作用。并且,脂肪细胞因子也存在抵抗作用,例如瘦素作为治疗肥胖的药物最主要的障碍是肥胖个体体内存在瘦素抵抗。除此之外,在人群实验中脂肪细胞因子的治疗也存在一定副作用风险。例如,高含量的脂联素水平通常认为与胰岛素敏感性和2型糖尿病的低风险密切相关,但脂联素对个体死亡率的影响尚未在大型遗传研究中得到证实[85]。这些原因限制了脂肪细胞因子在代谢性疾病治疗中的运用。因此,从动物结果到人群治疗的转换需要更多实验探究及时间的检验,加快针对脂肪细胞因子的研究对糖尿病等代谢性疾病的治疗具有重大意义。

Fig.2 The regulation of adipokines on metabolism Effects of adipokines on muscle and liver metabolism. Red and blue arrows represent upregulation and downregulation,respectively. FFA, free fatty acids; TG, triglycerides; IR, insulin resistance
猜你喜欢
恋爱婚姻家庭·养生版(2018年6期)2018-08-01
特别健康·下半月(2018年2期)2018-04-24
江苏农业科学(2017年17期)2017-11-15
中国当代医药(2016年28期)2016-12-20
科学中国人(2016年9期)2016-11-04
医学信息(2016年9期)2016-05-14
健康博览(2015年3期)2015-07-03
中国现代医生(2015年4期)2015-03-11
商情(2009年17期)2009-09-23
