
126111例新生儿串联质谱遗传代谢病筛查结果及基因突变情况分析
2022-09-02 01:54王欣荣刘艳秋官慧珍王枫黄淑晖杨必成
江西医药 2022年7期
王欣荣,刘艳秋,官慧珍,王枫,黄淑晖,杨必成
(江西省妇幼保健院医学遗传中心江西省出生缺陷防控重点实验室,南昌 330006)
遗传代谢病由英国医生Garrod在1908年首次提出,通常是由于酶、辅酶或转运蛋白的缺陷而导致底物积累或下游产物不足所致疾病的统称,涉及脂肪酸、氨基酸、有机酸、尿素等物质的代谢等[1]。虽然遗传代谢病单病种发病率较低,但总体发病率并不低,有研究显示每3795名新生儿中就有一人患,随着筛查检测和诊断的技术进步,更多的遗传代谢病被发现[2]。一些遗传代谢病可能会出现危及生命的急性症状,使临床早期发现这种疾病。但遗传代谢病的大多数症状是非特异性的,包括嗜睡、呕吐、特征性气味、酸中毒和大脑发育迟缓等[3],如果不及早诊断和治疗,其中大多数可能会导致智力障碍或发育迟缓等严重的永久性后遗症,在某些情况下还会死亡[4]。因此针对遗传性代谢性疾病,早期筛查早发现是关键环节,串联质谱技术比传统的分析更灵敏、特异、可靠和全面,一次测试可以同时测定和定量多种氨基酸和酰基肉碱,可以同时筛查检测多种遗传代谢病[5]。江西省妇幼保健院江西省新生儿疾病筛查诊治中心于2016年在江西省内率先应用串联质谱技术进行新生儿遗传代谢病筛查[6],为了解串联质谱新生儿遗传代谢病患病率及基因突变情况,本研究回顾性分析江西省妇幼保健院2016年1月至2021年12月出生的126111例活产新生儿串联质谱遗传代谢病筛查情况,报道如下。
1 资料与方法
1.1 一般资料 研究对象为江西省妇幼保健院2016年1月至2021年12月出生的126111例进行串联质谱遗传代谢病筛查新生儿,筛查前均获得家属知情同意。
1.2 标本采集 采集出生72小时后并充分哺乳至少8次以上的新生儿足跟血,采血部位为足跟内外侧缘,采血深度不超过2 mm,早产儿或低体重儿应适当浅些,采血前用75%的乙醇消毒足跟,用无菌棉球擦去第一滴血,连续采集3~4个直径不小于8 mm、圆形均匀血斑于S&S903滤纸片,悬空室温2~3 h晾干后,置于密封袋封口,2~8℃保存,标本应于5个工作日送至筛查中心,初筛阳性者召回复查,复查阳性结合临床招生指标为可疑遗传代谢病患者进一步基因诊断。
1.3 实验仪器及方法 取1个滤纸干血斑点(直径3.0 mm),置于96微孔U型板中,每孔加入含氨基酸和酰基肉碱同位素内标的萃取液(甲醇:水,80∶20,v/v,含0.01%甲酸)100μL,孵育振荡45 min,然后转移上清液80μL至另外一个96微孔板中,取20μL进样,进行串联质谱分析(ABsciex 3200)。
1.4 初筛阳性复查 针对初筛过程中存在的阳性新生儿,则召回相应新生儿,对其进行再次采血进行串联质谱遗传代谢病筛查,如果召回复查结果仍高于正常值,则为可疑阳性,进一步结合临床表现和其他生化指标,对其进行突变基因分析。
2 结果
2.1 126111例新生儿串联质谱遗传代谢病筛查结果分析 126111例新生儿串联质谱遗传代谢病筛查初筛阳性3657例,阳性率2.9%;召回3498例,召回率95.66%,最终确诊65例,总体患病率为1/1940。见表1。

表1 126111例新生儿遗传代谢病筛查结果
2.2 脂肪酸代谢障碍疾病基因诊断结果 24例脂肪酸代谢异常患儿进行了基因诊断。其中9例原发性肉碱缺乏症患儿进行了基因诊断,基因型以c.51C>G(p.F17L)和c.1400C>G(p.S467C)常见;5例母源性肉碱缺乏症患儿中基因型以c.1400C>G(p.Ser467Cys)常见;5例短链酰基辅酶A脱氧酶缺乏症的基因型以(c.1031A>G)(p.E344G)常见。见表2。

表2 脂肪酸代谢障碍疾病基因诊断结果
2.3 氨基酸代谢障碍疾病基因诊断结果 16例氨基酸代谢障碍患儿进行了基因诊断。9例高苯丙氨酸血症患儿进行了基因诊断,基因型以c.1174T>A常见;3例四氢生物蝶呤缺乏症患儿基因型以c.259C>T常见。见表3。
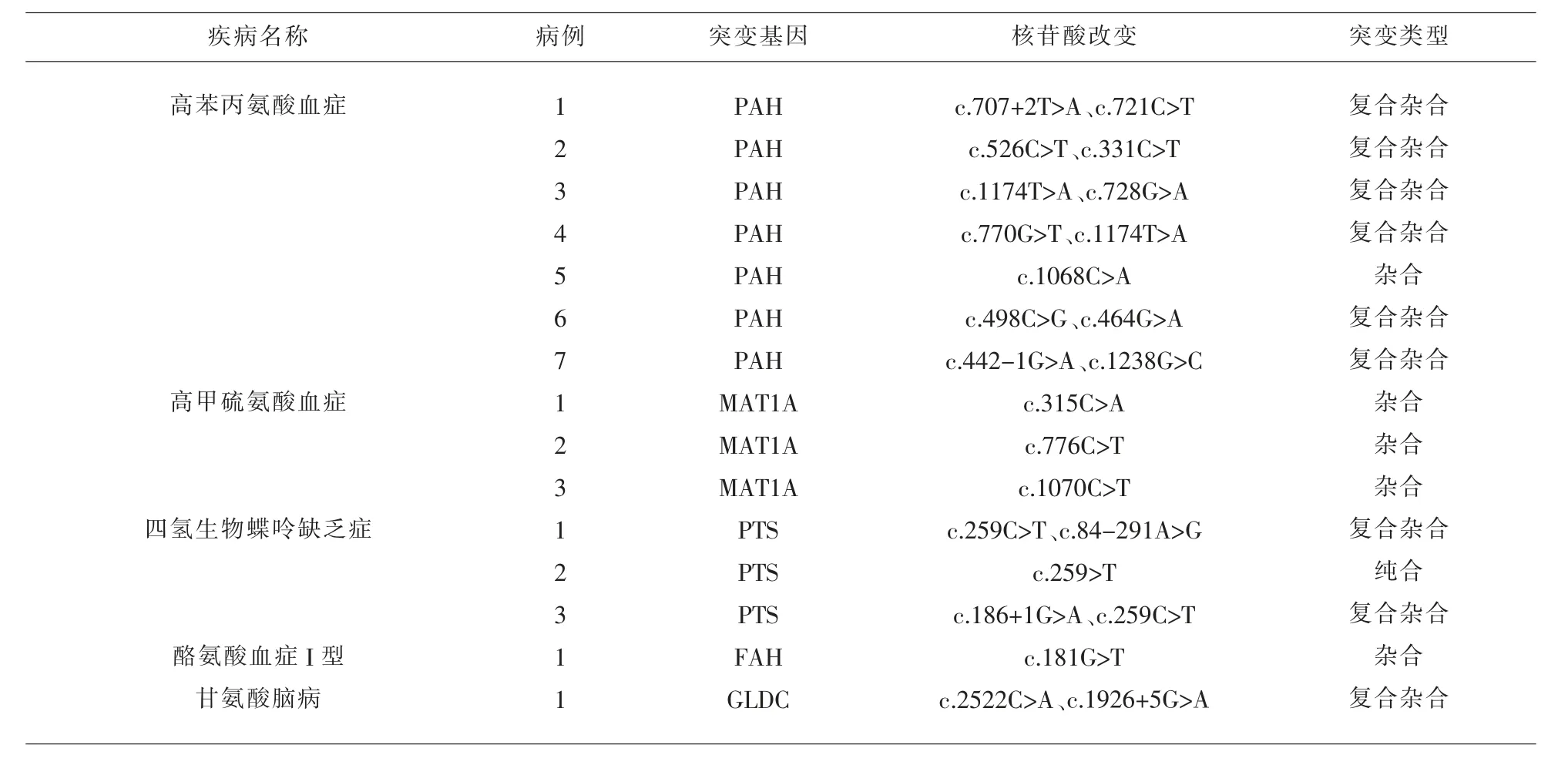
表3 氨基酸代谢障碍疾病基因诊断结果
2.4 有机酸代谢障碍疾病基因诊断结果 4例有机酸代谢障碍患儿进行了基因诊断。其中2例甲基丙二酸血症患儿进行了基因诊断,1例突变类型为MUT基因C.323G>A纯合突变,另1例为MMAB基因c.571C>T、c.539C>G复合杂合突变;1例3甲基巴豆酰辅酶A羧化酶缺乏症患儿进行了基因诊断,为MCCC1基因c.1570G>C、c.601C>T复合杂合突变;1例母源性3甲基巴豆酰辅酶A羧化酶缺乏症为MCCC1基因c.1032T>A杂合突变。
2.5 尿素循环障碍疾病基因诊断结果 5例有尿素循环障碍患儿进行了基因诊断。3例希特林蛋白缺乏症患儿基因诊断,均为SLC25A13基因的复合杂合突变,2例基因型为c.852_855del,另1例基因型为c.855delTATG和IVS16ins3kb;1例精氨酸琥珀酸尿症患儿为ASL基因外显子5的c.331C>T(p.R111W)和外显子9的c.655+1_655+2insT;1例鸟氨酸氨甲酰转移酶缺乏症患儿为OTC基因c.275G>A p.R92Q半合子错义突变。
3 讨论
遗传代谢病患者若得不到不及时诊治,常可致残,甚至危及生命,既严重影响出生人口素质,又给脱贫攻坚带来严峻挑战[7]。早期诊断是进行及时处理、挽救生命、避免或减少严重并发症及神经系统损伤的关键,也是进一步进行遗传咨询或产前诊断的基础。在新生儿期进行遗传代谢病筛查,对出生缺陷的防控具有重要意义。
1990年美国杜克大学Dr Millington等首次将串联质谱技术应用于新生儿遗传代谢病筛查,该技术能在2~3分钟内对一个标本进行几十种小分子代谢物进行检测,通过对小分子代谢产物进行分析可以对40余中新生儿遗传代谢病进行筛查检测,使得新生儿遗传代谢病筛查提高到一个新的水平。新生儿疾病筛查对于疾病的三级预防具有重要价值[8],随着医学技术的不断进步,串联质谱技术已逐步应用于新生儿遗传代谢病筛查中,江西省妇幼保健院江西省新生儿疾病筛查诊治中心于2016年在江西省内率先应用串联质谱技术进行新生儿遗传代谢病筛查。本文分析了江西省新生儿筛查中心2016年1月到2021年12月的126111例新生儿用串联质谱技术的筛查结果。结果表明,126111例新生儿中初筛阳性3657例,阳性率2.9%;确诊65例,总体患病率为1/1940,低于济南市1/1178[9]、石家庄市1/1259[10],但高于广州市1/2451[11]、苏州市1/2910[12],发病率前三位分别为原发性肉碱缺乏症、高苯丙氨酸血症、希特林蛋白缺乏症。由此可见不同地区的新生儿遗传代谢病的发病率存在明显差异,同时证明江西省脂肪酸代谢障碍特别是原发性肉碱缺乏症发病率较高。研究表明与未接受筛查的人群相比,串联质谱技术的普遍筛查显著节省了医疗成本,同时有助于降低儿童死亡率和减少长期残疾[13]。
综上所述,串联质谱技术的应用显著提高了新生儿疾病筛查的效率。对于被诊断为遗传代谢病的患儿,如果实施早期诊断和治疗,可以防止机体组织器官发生不可逆的损伤,避免患儿发生智力障碍、严重的疾病甚至死亡等严重的临床后果。患儿可得以早期诊断、早期治疗,从而减少出生缺陷发生,降低死亡率。
猜你喜欢
当代医药论丛(2022年17期)2022-10-09
少男少女·教育管理(2022年3期)2022-05-12
现代仪器与医疗(2022年1期)2022-04-19
北京航空航天大学学报(2021年4期)2021-11-24
现代仪器与医疗(2021年2期)2021-07-21
大自然探索(2020年5期)2020-06-19
农家科技(2019年5期)2019-07-08
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
作文周刊·小学一年级版(2016年11期)2016-05-10
