
Mechanism for development of malnutrition in primary biliary cholangitis
2022-08-26 09:49VasiliyIvanovichReshetnyakIgorVeniaminovichMaev
World Journal of Meta-Analysis 2022年3期
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Abstract Primary biliary cholangitis (PBC) is a chronic cholestatic liver disease that is associated with impaired biliary excretion processes. Along with the development of cholestasis, there is a deficient flow of bile acids into the intestinal lumen causing malnutrition (MN) that is manifested in deficiencies of both macro- and micronutrients. The mechanism for development of trophological insufficiency is multifactorial. However, the trigger of MN in PBC is impaired enterohepatic circulation of bile acids. The ingress of bile acids with a detergent effect into the general bloodstream, followed by elimination via the kidneys and skin, triggers a cascade of metabolic disturbances, which leads to the gradual development and progression of calorie MN. The latter gradually transforms into protein-calorie MN (PСM) (as marasmus) due to the insufficient entry of bile acids into the duodenum, which is accompanied by a decrease in the emulsification, hydrolysis, and absorption of fats and fat-soluble vitamins, as well as disturbance of intestinal motility and bacterial overgrowth. Fat-soluble vitamin deficiencies complement P СM with vitamin and mineral MN. The development of hepatocellular failure enhances the progression of PСM due to the impaired protein synthetic function of hepatocytes in the advanced stage of PBC, which results in deficiency of not only the somatic but also the visceral pool of proteins. A mixed PСM form of marasmus and kwashiorkor develops. Early recognition of energy, protein, micronutrient, and macronutrient deficiencies is of great importance because timely nutritional support can improve liver function and quality of life in patients with PBC. In this case, it is important to know what type (energy, proteincalorie, vitamin, and vitamin-mineral) and form (marasmus, marasmuskwashiorkor) of MN is present in the patient and how it is associated with the stage of the disease. Therefore, it is recommended to screen all patients with PBC for MN, from the early asymptomatic stage of the disease in order to identify and avoid preventable complications, such as fatigue, malaise, performance decrement, sarcopenia, osteoporosis, and hepatic encephalopathy, which will be able to provide appropriate nutritional support for correction of the trophological status.
Key Words: Primary biliary cholangitis; Мalnutrition; Сalorie; Protein-calorie; Vitamin-mineral malnutrition; Мarasmus and marasmus-kwashiorkor malnutrition
lNTRODUCTlON
Malnutrition (MN) is common in patients with primary biliary cholangitis (PBC). MN accompanying this disease worsens its course, prognosis, and quality of life in a patient, negatively affects the outcome of the disease, and is most often recognized only in its later stages[1]. The etiology and pathophysiology of MN are multifactorial in PBC[2]. Impaired biliary excretion processes in PBC, which are accompanied by cholestasis and decreased hepatocyte function, affect the metabolism of both macronutrients and micronutrients and depends on the stage of the disease. For the timely diagnosis and correction of the abnormal trophological status, it is important to understand when, at what stage of the disease, and by what mechanisms, calorie, protein, vitamin, and mineral MN develop in PBC patients. To improve treatment results in patients with PBC, it is necessary to pay attention to the development of MN in them and to its prevention and treatment even in the early stages of the disease. The advanced, endstages of PBC are accompanied by an imbalance between catabolism and anabolism, with the predominance of the former over the latter. The goals of nutritional therapy for patients with cholestatic liver disease are improvement of anabolic processes for valuable liver regeneration, prevention, and correction of malnutrition as well as to avoid and/or treat related complications of liver disease. It is very important to focus not only on the specific signs of the disease but also on the assessment of the nutritional status in patients during their initial examination. At the same time, the features and mechanisms of metabolic disorders should be taken into account in different stages of PBC in order to timely recognize MN and its correction during basic treatment, taking into account currently known scientific data.
DEFlNlTlON OF MALNUTRlTlON
The Russian literature lacks the generally accepted term to define the nutritional status[3]. MN (synonyms: Protein-calorie, nutritional insufficiency, trophological insufficiency, malnutrition) is a pathological condition caused by a discrepancy between the intake and consumption (imbalance) of organic nutrients, calories, macronutrients, and micronutrients, leading to weight loss, a measurable negative change in the component composition of the body, which is accompanied by its dysfunction and a poorer clinical outcome[4,5]. MN is defined by the World Health Organization as the result of insufficient intake or absorption of nutrients needed to support growth and to prevent chronic or acute diseases and is often characterized by growth retardation, wasting, being underweight, and micronutrient deficiencies[6]. MN is accompanied by weight loss, lower physical performance, and worse health and causes serious metabolic disorders, immunosuppression, and endocrine dysfunctions[7,8].
PREVALENCE OF MALNUTRlTlON lN PATlENTS WlTH LlVER DlSEASE
It is very difficult to estimate the true prevalence of MN for the following reasons[5]: Physicians’ extremely low attention to the trophological status; difficulties in assessing MN; and the masking of muscle tissue loss in the presence of excess fat mass and fluid retention.
About 2 billion people in the world experience various types of MN[9]. Secondary (endogenous) MN is noted in patients with various diseases. Studies indicate that MN is observed in 20%-80% of patients with liver disease, according to the clinical stage of the disease[10]. Almost any chronic illness can cause progressive weight loss. A study by Carvalho and Parise[11] has shown that as many as 75% of patients with chronic liver disease have varying degrees of MN. Hyponutrition and sarcopenia are common in patients with chronic liver disease and are associated with an increased risk for decompensation and infections as well as are frequently an independent risk factor for death in these patients and worse treatment outcomes after liver transplantation[2,12]. It is important to note that the incidence of trophological insufficiency increases in these patients as the disease progresses. In the 1990s studies[13,14], evaluation of the nutritional status in patients with different etiologies of liver cirrhosis and with various degrees of liver failure came to the consensus that MN was recognized in all types of cirrhosis[15] and, according to various authors, it ranged from 40% to 100%[16-19]. There is a high prevalence of MN in individuals with decompensated liver cirrhosis. The prevalence of MN is 46% in patients classified as Child-Pugh A cirrhosis and 84% and 95% in those classified as Child-Pugh B and C, respectively[11]. MN cases can be as much as 100% in patients on the waiting list for liver transplantation[20,21].
PBC AND MALNUTRlTlON
MN develops in PBC patients with and without established cirrhosis[22,23]. According to Wickset al[22], MN is detected in 33% of patients with different stages of PBC. Primary biliary cholangitis (PBC; ICD-10 K.74.3; ICD-11 (beta draft) DB37.2) is a disease, formerly (until 2015) known as primary biliary cirrhosis (PBC)[24], is the chronic, progressive autoimmune cholestatic liver disease proceeding with epithelial destruction, necrosis, and apoptosis, mainly affecting the intralobular and septal bile ducts, which eventually leads to liver cirrhosis[25,26]. PBC is characterized by T-cell-mediated destruction of epithelial cells that line the small intrahepatic bile ducts[27]. This leads to ductulopenia and persistent cholestasis to develop end-stage cirrhosis with hepatocellular failure[26].
Early-stage disease may be asymptomatic or have nonspecific symptoms, such as weakness, fatigue, reduced performance, anorexia/hyporexia, and malaise, which can be easily confused with other conditions. During this period, MN caused by the disease itself is generally invisible since there are no significant damage to the liver cells involved in metabolic processes. In early-stage PBC, there is a slight decrease in energy consumption, which does not lead to clinically pronounced protein-calorie MN, but there is already a potentially modifiable MN during this period[28-30].
As cholestasis progresses, excess bile acids have a chronic (continuous) aggressive effect on the liver parenchyma, which is manifested by the development of gradually progressing hepatocellular failure. The trophological status of patients with PBC also decreases as the disease progresses, which is partly due to a significant decrease in energy consumption[22]. Patients with advanced PBC develop liver cirrhosis that may be accompanied by ascites, portal hypertension, esophageal/gastric variceal hemorrhage, and hepatic encephalopathy[31]. Portal hypertension can develop in patients with cholestatic liver disease before cirrhosis is established[32,33]. There is almost a direct relationship between the severity of liver disease and the degree of MN[30]. In this case, the state of nutrition is disturbed secondarily to the symptoms of the disease[33]. Severe protein-calorie MN more frequently develops and is observed in advanced and end stages of PBC, generally in patients who have been suffering from this disease for more than one decade[29] and when there is a 25% or less decline in the total number of functioning hepatocytes[34]. Trophological insufficiency becomes more easily detectable when the patients with PBC develop severe cirrhosis with ascites.
THE PATHOGENESlS OF MALNUTRlTlON lN PBC
The causes and mechanisms leading to MN and weight loss in patients with PBC are multifactorial and can be divided into three groups: insufficient intake of nutrients; abnormalities in digestion and absorption (maldigestion and malabsorption syndromes); and increased metabolic rate (accelerated catabolism).
Insufficient intake of nutrients in patients with PBC
In early-stage PBC, the trophological changes are associated with elevated plasma bile acid levels in these patients, which gives rise to an early and, most commonly, the only for several months or even years pathognomonic symptom of the disease, such as local or diffuse (extension), moderate or pronounced (degree), and persistent or transient (duration) skin itching[26,35]. The cause of skin itching is the epidermal deposition of bile acids that are abundant in the blood of patients with PBC even in the asymptomatic and early stages of the disease, long before developing jaundice. In this case, all fractions of conjugated bile acids increase in the blood.
In response to excess plasma bile acids in PBC patients, whose body tries to remove toxic bile acids having detergent properties from the general circulation through the kidneys and skin. In this case, 50%-85% of bile acids that are unconjugated with glycine or taurine are detected in the skin, and less than 20% of bile acids are found as sulfoesters[25,26,36]. Itching is more marked at night and frequently enhanced in contact with tissues as well as in warmth. Itching is not relieved by symptomatic (antihistamine, sedative) medications; it often causes excruciating insomnia, emotional changes, anxiety, and depression[24]. All this leads to decreased appetite and insufficient intake of food ingredients, which is accompanied by an increase in glycogenolysis and by a reduction in glycogenogenesis.
Glycogenolysis, a biochemical process of breaking down glycogen into glucose, occurs primarily in the liver and muscles. The main purpose of glycogenolysis is to keep blood glucose levels stable to provide the body with energy. Due to its glycogen stores and glycogenolysis processes, the liver provides up to 75% of the body’s need for glucose as the primary substrate quickly used to replenish energy.
Glycogenogenesis is a metabolic pathway that synthesizes glycogen from glucose. This process takes place in all tissues; however, it occurs mainly in the liver and muscles. The starting point for glycogenogenesis is glucose-6-phosphate that can be obtained from glucose in the reaction catalyzed by glucokinase in the liver and by hexokinase in the muscles. Liver glycogen is known to be used as an energy material by all tissues and organs. At the same time, glycogen in muscles is employed by them as an energy material exclusively for their own needs.
Greenet al[37] indicated that even in the early stages of PBC, glycogen stores gradually reduce in the liver, which is associated with an increase in glycogenolysis and a decrease in glycogenogenesis. The authors have convincingly shown that glucokinase activity in PBC patients reduces significantly (down to zero), which suggests that liver glycogen production decreases[37]. At the same time, hexokinase (performs phosphorylation of hexoses) that is responsible for glycogen synthesis mainly in the muscles significantly increases during this period in PBC patientsvshealthy individuals[37]. At the same time, hexokinase (that produces phosphorylation of hexoses), which is responsible for glycogen synthesis mainly in the muscles, significantly increases during this period in PBC patientsvshealthy individuals[37].
Excruciating insomnia, emotional changes, anxiety, and depression, which develop even in the early stages of the disease, contribute to a decline in glycogen stores and to gradually progressing energy deficiency (a reduction in the level of glucose used as an energy substrate) with the clinical manifestations of obvious weakness, rapid fatigue, and decreases in performance, functional status, and quality of life in PBC patients[24,33,35,38-40]. Moreover, the asthenic syndrome in patients with PBC is more pronounced than in those with other chronic liver diseases[24]. There is evidence that an important role in the mechanism of fatigue development is played by aromatic amino acids, such as tyrosine, phenylalanine, and tryptophan, which are abundant in the blood of patients with PBC[39,41,42].
And so, in early-stage PBC, an imperceptible trophological change occurs as calorie MN that manifests itself only as general weakness and/or reduced working capacity for a fairly long time[40,43,44].
The developing impairment of biliary excretion processes (accumulation of blood bile acid) in patients with PBC even in the asymptomatic and early stages of the disease contributes to the development of calorie MN, which requires that higher-calorie foods be included in the diet of these patients.
Even slight nutrient deficiencies accompanied by a gradual increase in glycogenolysis and a decrease in glycogenogenesis leads to the inclusion of compensation mechanisms. The latter are intended to protect higher energy-consuming vital organs (the brain, myocardium, erythrocytes,etc.) from energy deficiency by redistributing plastic and energy resources[5]. This brings about the mobilization of energy resources of adipose tissue and the consumption of fatty acids as an energy material. Fatty acids become important substrates for energy production. Due to the acceleration of fatty acid β-oxidation processes, there is a progressive decline in fat stores in patients with PBC[45,46]. The activation of these processes occurs as cholestasis progresses.
Along with this, patients with PBC are observed to have elevated levels of palmitic and oleic fatty acids[37]. The latter are the main components of biliary phospholipids (Figure 1) that are involved in the formation of micellar and lamellar structures consisting of phospholipids, cholesterol, and bile acids[47]. In patients with PBC, the plasma levels of palmitic and oleic acids as well as phospholipids and cholesterol increase even in the early stages of the disease and are aimed at neutralizing the detergent effect of excess bile acids entering the general circulation as cholestasis progresses[26,32]. In PBC patients, cholesterol, phospholipids, and palmitic and oleic acids can enter the general circulation due to an increase in their synthesis in the liver and to the entry of bile components into the blood as a consequence of progressive cholestasis.
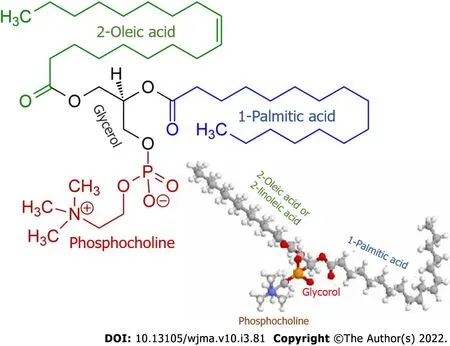
Figure 1 Chemical structure of phosphatidylcholine containing palmitic and oleic fatty acids.
The higher synthesis of phospholipids requires an increased amount of not only palmitic and oleic fatty acids but also orthophosphate. In this connection, even in the early stages of the disease, the plasma of patients with PBC displays the moderately enhanced activity of the hepatic fraction of alkaline phosphatase and 5’-nucleotidase, which indicates changes in phosphorus metabolism[26]. These enzymes are involved in the hydrolysis of phosphomonoesters to yield orthophosphate that is essential as the main component for the biosynthesis of phospholipids that in turn are required to neutralize the increased content of plasma bile acid levels in patients with PBC.
In patients with PBC, the long-term elevated plasma levels of cholesterol in response to the increase in its synthesis in the liver can give rise to xanthelasmas, single or multiple, pale-yellow formations that are slightly raised above the skin. In these patients, the increased levels of cholesterol as well as those of phospholipids are aimed at neutralizing the detergent effect of bile acids entering the general circulation. At the same time, despite the increase in their total plasma cholesterol, the patients with PBC were found to have mild hepatic steatosis and a low risk for atherosclerosis and cardiovascular events[48].
The developing impairment of biliary excretion processes (accumulation of blood bile acids) in patients with PBC even in its early stages causes fat metabolic changes that are aimed at compensating for energy deficit (accelerated fatty acid β-oxidation) and at neutralizing the detergent effect of excess bile acids (the increased synthesis of phospholipids and cholesterol). Therefore, standard foods are generally well tolerated by PBC patients who do not require a low-cholesterol diet in the early stages of the disease. Their diets can include foods that are high in phosphorus to maintain sufficient synthesis of phospholipids. Low-fat diets to reduce xanthelasmas have been recognized to be unsuccessful and even harmful[49].
Abnormalities in digestion and absorption (maldigestion and malabsorption syndromes)
Intrahepatic cholestasis in PBC is a multifactorial process that is associated with damage to subcellular structures in the epithelial cells of the intrahepatic bile ducts and with changes in bile acid metabolism due to impaired bile excretion. Insufficient entry of bile acids into the intestinal lumen in patients with PBC tends to decrease the rate of fat hydrolysis processes and to inadequately absorb fats and fatsoluble vitamins (A, D, E, and K) in the small intestine. This contributes to the progression of MN due to steatorrhea (fecal excretion of more than 7 g of fat per day) and to the gradual development of vitamin and mineral deficiencies[25,26]. The severity of steatorrhea correlates with lower bile acid production and concentrations (r= 0.82;P< 0.0001), elevated serum bilirubin levels (r= 0.88;P< 0.001), and late histological stages of PBC (P< 0.005)[50]. All patients with serum total bilirubin levels greater than 4.5 mg/dL have severe steatorrhea (the fecal fat excretion is greater than 25 g/d)[26,50].
The mechanism of steatorrhea development is associated with insufficient fat emulsification owing to the reduced ingress of bile acids into the intestinal lumen[51]. In this case, the processes of fat hydrolysis by pancreatic lipases are not disrupted. The results obtained by Roset al[52] showed that pancreatic function was generally preserved and does not cause steatorrhea in PBC. In patients with PBC, the serum activity of alkaline phosphatase does not correlate with the severity of steatorrhea, and the pancreatic amylase is in the normal range[52]. Fat emulsification is required to increase the area of contact of the substrate with lipase enzymes. A decrease in the processes of fat emulsification leads to the lower rate of hydrolysis of fats, which results in their incomplete digestion, when moving along the intestine, and contributes to the gradual development of steatorrhea.
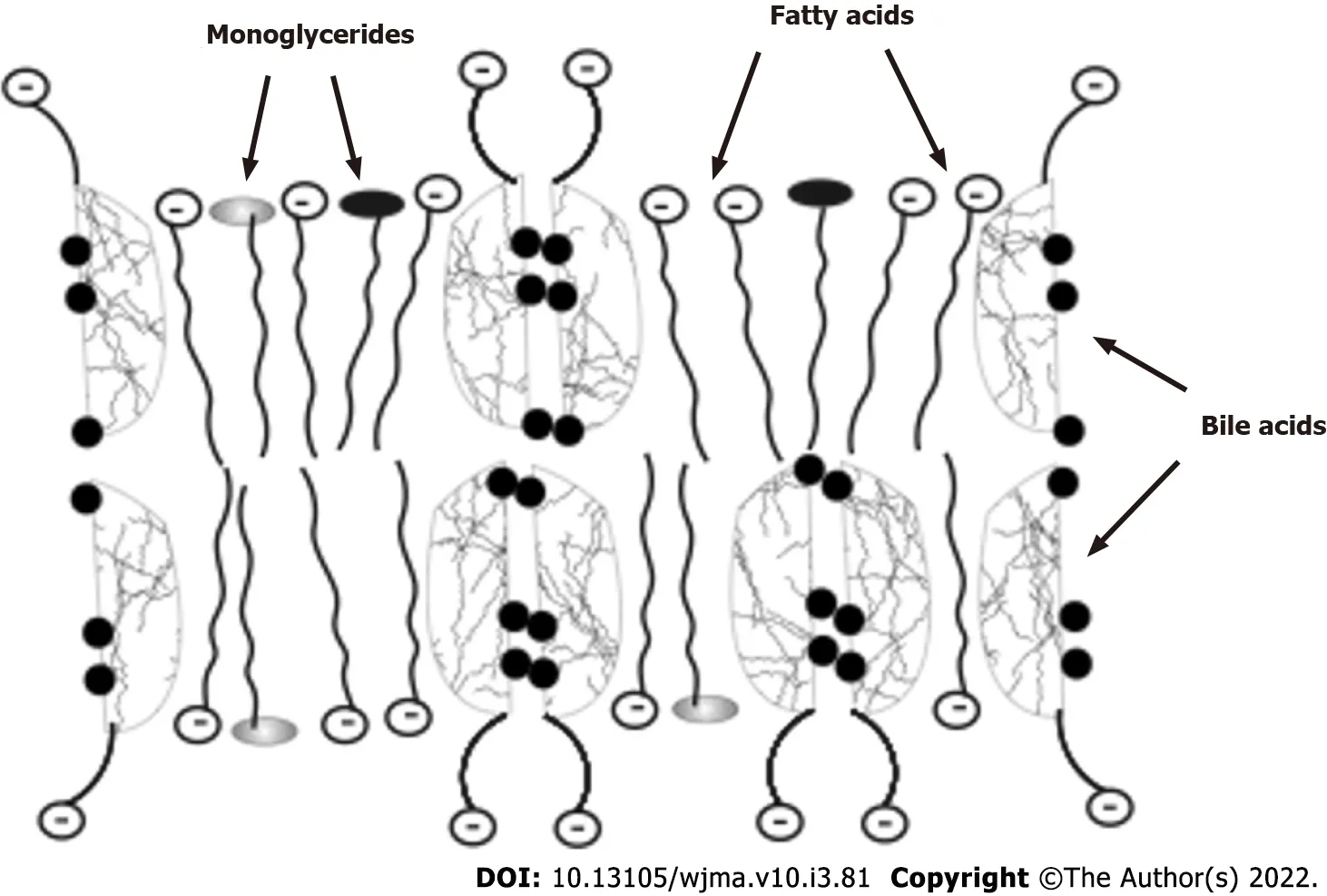
Figure 2 Schematic diagram of the composition of lipoid-bile acids complexes formed in the small bowel.
In addition to the emulsification of fats, bile acids are involved in the absorption of hydrolyzed fats and fat-soluble vitamins. Fatty acids and monoglycerides, which are formed from neutral fats and phospholipids, with the participation of bile acids and under the action of lipases are absorbed by enterocytes as an emulsion of lipoid-bile acid complexes in the upper small intestine (Figure 2). Being potent detergents, bile acids form micellar or lamellar structures with fatty acids and monoglycerides for absorption by enterocytes[25,26]. The complexes disintegrate inside the enterocytes. Fatty acids with monoglycerides remain in the enterocytes (used by a cell as a building, energetic material or packed into chylomicrons), while bile acids come back into the intestinal lumen and take part in the emulsification of fats and in the formation of new lipoid-bile acids complexes for delivery of fatty acids, monoglycerides, and fat-soluble vitamins to the enterocytes. While moving through the small intestine, bile acids are able to participate 4-6 times in the delivery of fatty acids and monoglycerides into the enterocytes[53]. Thus, insufficient amounts of bile acids in PBC interfere with the absorption of fats and fat-soluble vitamins.
Intestinal bile acid deficiency not only impairs fat emulsification and the decreased absorption of fatty acids, monoglycerides, and fat-soluble vitamins, in patients with PBC[52] but also leads to intestinal microbiome dysbiosis[54]. DiBaiseet al[55] suggested that dysbiosis also plays a significant role in the development of steatorrhea in patients with PBC and that bacterial overgrowth should be obligatorily assessed in these patients.
Since the insufficient entry of bile acids into the intestine is one of the first signs of the disease, even in its early stages, patients with PBC can be found to have fecal matter of incompletely digested fats, one of the signs of steatorrhea. As the disease progresses and steatorrhea develops, most patients have semiliquid stools up to diarrhea of varying severity. With this, some patients with PBC are observed to have constipation. The latter can be attributed to a certain change in the gut microbiome and an insufficient effect of a small amount of bile acids on intestinal motility.
Steatorrhea in the presence of gradually and imperceptibly developing calorie MN leads to the development of slowly progressive weight loss in patients with PBC. Mid-arm circumference, triceps skinfold thickness, and dual energy X-ray absorptiometry-estimated % fat decreased significantly with disease progression (P< 0.001) and especially when liver cirrhosis with ascites is established[22].
The development of slowly progressive weight loss can be facilitated by the use of certain drugs. Thus, cholestyramine used to relieve itching can cause abdominal distention, constipation, or diarrhea, which suppresses, restricts, and disrupts food intake, resulting in inadequate intake of food ingredients and in higher energy deficiency[32].
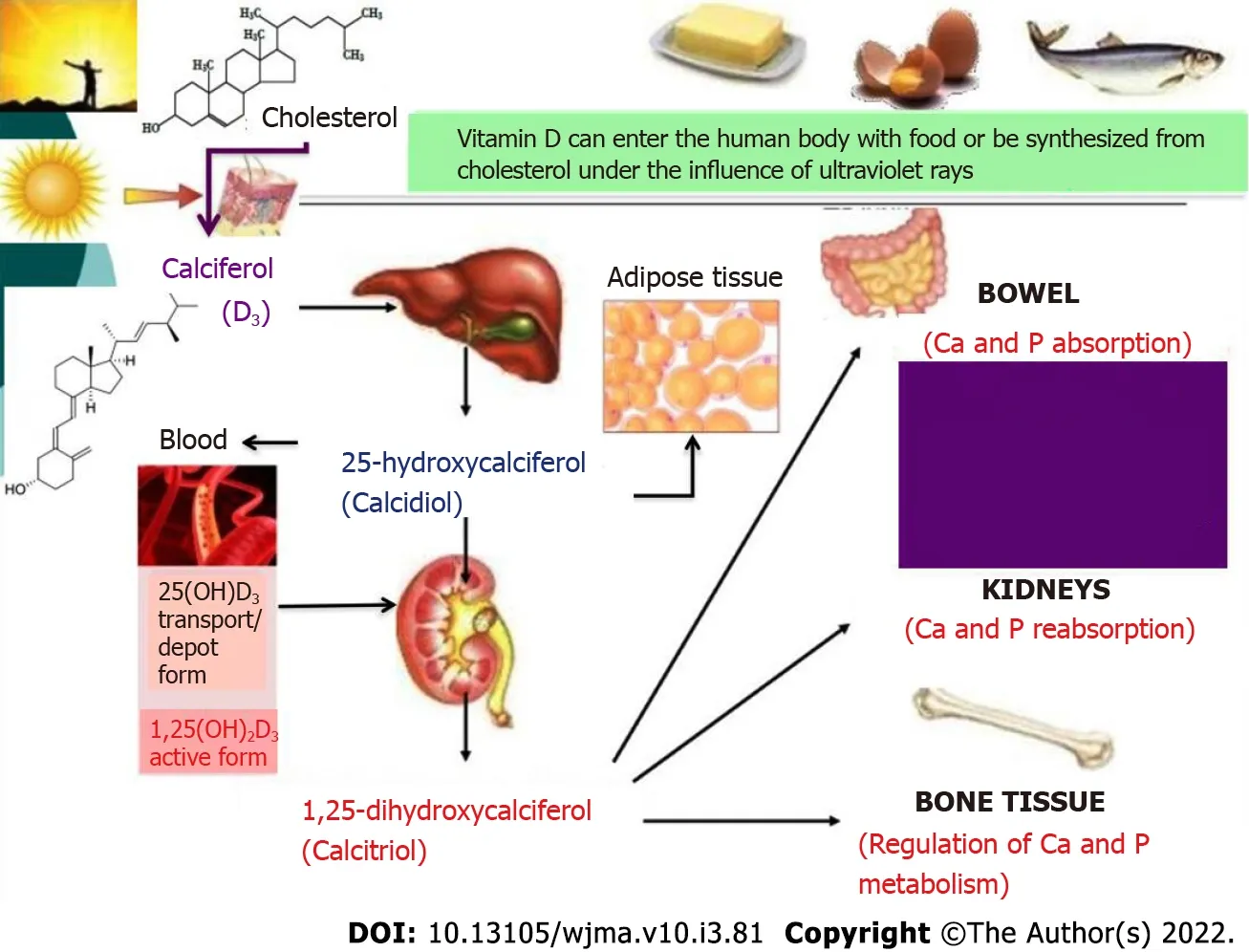
Figure 3 Metabolism of vitamin D and its involvement in the metabolism of phosphorus and calcium.
The developing impairment of biliary excretion processes (insufficient flow of bile acids into the duodenum) in patients with PBC even in its early stages contributes to the development of slowly progressive weight loss, which requires the prescription of ursodeoxycholic acid preparations and the incorporation of high-calorie foods for their diets. Since these patients eat less during meals due to early satiety, it is possible to recommend smaller, more frequent higher-calorie meals[28,29]. At the same time, fats should not be restricted in their eating patterns. Edible fats should be restricted only if the patients have obvious steatorrhea or severe nausea or symptoms of indigestion when consuming fats. However, it should be borne in mind that foods that have no or low fat and/or triglycerides containing the average fatty acid chain length are generally better tolerated. Therefore, it is important to individually assess the patients’ tolerances to different fats and to make appropriate recommendations.Vitamin and mineral deficiencies:Bile acids play an important role in absorbing the fat-soluble vitamins A, D, E, and K from the intestines. Bile acids can include fat-soluble vitamins in the lipoid-bile acid complexes and thus transport them into the enterocyte. Insufficient entry of bile acids in the intestine in PBC leads to a decrease in the absorption of fat-soluble vitamins and to the development of vitamin deficiencies[56]. Deficiency of vitamins A, D, E, and K was identified in 33.5%, 13.2%, 1.9%, and 7.8% of PBC patients, respectively[56].
Despite the fact that insufficient ingress of bile acids in PBC occurs even in its early stages, fat-soluble vitamin deficiencies are more frequently manifested at the stage of frank cholestasis with pronounced signs of the disease or in the stage of cirrhosis development. The ability of fat-soluble vitamins to accumulate in significant quantities and to be stored in the liver and adipocytes as well as that of some of them to be synthesized in the body cause fat-soluble vitamin deficiencies to generally not develop in early-stage PBC. Thus, vitamin D is synthesized in the skin from cholesterol upon exposure to ultraviolet rays, whereas vitamin K is synthesized by the intestinal microflora. But as the disease progresses and hepatocellular failure develops, there is deficiency of these fat-soluble vitamins since they are metabolized in the liver.
Vitamin D takes an active part in phosphorus and calcium metabolism. Dietary (edible) vitamin D and vitamin D3(calciferol) that is newly synthesized by ultraviolet radiation from cholesterol are inactive forms of this vitamin. In the liver, it is hydroxylated to 25-hydroxycalciferol (calcidiol) that can accumulate and be stored in the liver and adipose tissue. The serum concentration of 25-hydroxycalciferol is considered the most reliable indicator of the total metabolism of vitamin D; therefore, this indicator can be used to determine the body’s supply of this vitamin[25,26]. When blood calcium decreases, there is an increase in the synthesis of parathyroid hormone that stimulates the renal hydroxylation of calcidiol to 1,25-dihydroxycalciferol (calcitriol), the active form of vitamin D, which is also involved in the regulation of metabolism of calcium and phosphorus: it increases their absorption in the intestine, their reabsorption in the renal tubules, and regulates the exchange of calcium and phosphorus in the bones (Figure 3).
In PBC, as hepatocellular failure develops, there is a gradually progressive deficiency of calcidiol, precursor of the active vitamin D, leading to osteodystrophy accompanied by osteopenia. The latter is a recognized complication of cholestatic liver disease with a prevalence of 10% to 56% depending on the stage of the disease[57]. PBC is a condition that causes osteopenia more often than other chronic liver diseases[58-61], as clinically manifested by the development of signs of osteoporosis[62].
Osteoporosis:Osteoporosis is a systemic skeletal disease characterized by low bone mass and bone tissue microarchitectural deterioration, thus resulting in increased bone fragility and a higher risk for unmotivated fractures[63-65]. The prevalence of osteoporosis among patients with PBC ranges from 20% to 37% or more, which is higher than that in the general population (10%-11%)[59-61,66]. According to Lindoret al[32], the incidence of osteoporosis in PBC is 30%. Osteoporosis increases with liver disease progression[67].
The molecular mechanisms of osteoporosis in patients with PBC are associated with the impaired enterohepatic bile acid circulation, followed by the decreased concentration of bile acids in the small bowel and by malabsorption of fat-soluble vitamin D[68]. In PBC patients with severe cholestasis along with developed intestinal malabsorption of dietary vitamin D, calciferol is slowly converted to calcidiol in the liver as hepatic cell failure progresses[69] and due to those monooxygenases are competitively inhibited[70]. In these patients, the lower amount of calcidiol causes a decrease in the renal production of the active form of vitamin D, calcitriol. This results in an impairment of phosphorus and calcium metabolism, which gives rise to osteodystrophy[58,71,72] that can manifest itself as bone pain even in the early stages of PBC. The development of bone densitometry could estimate bone mass and assess the risk of fractures[58], which correlated with bone mineral density[64]. In this case, laboratory tests yield important information about the metabolic status of the bone. The serum level of calcium and phosphorus is usually slightly reduced in patients with PBC[58]. In Sherlock’s[51] opinion, impaired phosphorus and calcium metabolism in PBC patients is facilitated by steatorrhea; increased intestinal fat content can form insoluble soaps with calcium, preventing its further absorption by enterocytes. Reduced calcium absorption correlates well with increased fecal fat excretion and to a lesser extent with the severity of jaundice[73]. Genetic factors also play a role in the development of osteoporosis[74-76]. Bone X-ray and densitometry and morphological examination of a bone tissue biopsy specimen from a patient with PBC most commonly reveal the signs of osteoporosis[58]. In the later stages of the disease, there are pathological fractures of the vertebrae and ribs, less frequently those of pelvic bones and long bones[66].
In patients with PBC, long-term steroid therapy that accelerates and aggravates the development of osteoporosis can lead to clinically significant bone loss with a more than 2-fold increase in the risk of fractures[77]. Atraumatic fractures are especially dangerous in PBC patients who have undergone orthotopic liver transplantation and are treated with high-dose corticosteroids[62].
Glucocorticosteroids decrease the intestinal absorption of calcium by lowering the production of calcitriol (1,25-dihydroxyvitamin D3), by suppressing the tubular reabsorption of calcium in the kidneys, and by increasing its urinary excretion. A decrease in blood calcium levels leads to a compensatory increase in parathyroid hormone production and bone resorption. In addition, glucocorticosteroids directly increase the release of parathyroid hormone and suppress the function of osteoblasts by enhancing the activity of osteoclasts and indirectly inhibiting the formation of bone tissue, by suppressing the synthesis of testosterone in the gonads, and by reducing the generation of growth hormone, insulin-like growth factor 1 that is synthesized by the liver and stimulates bone collagen type 1 synthesis and osteoblastic function[58,78,79].
Thus, the pathogenesis of osteoporosis in patients with PBC is complex and multifactorial[58,62,80] and involves impaired vitamin D and K absorption and metabolism[81], magnesium ion deficiency, decreased intestinal absorption of calcium and its reabsorption in the renal tubules, increased bone resorption[82,83], elevated levels of bilirubin that inhibits osteoblast function[58,66,84], genetic predisposition[85], and adverse reactions of drugs, such as corticosteroids and cholestyramine, which are used to treat patients with PBC[86]. The development of osteoporosis is associated with the severity of the disease rather than its duration. Osteoporosis can affect quality of life and the course of the disease[58]. Deficiency of the active form of vitamin D (calcitriol) is a risk factor for osteosarcopenia[66,87].
Vitamin K:Vitamin K is required for the synthesis of coagulation factors VII, IX, and X and prothrombin in the liver[88-90]. During the early stages of PBC, vitamin K deficiency is generally absent. As malabsorption and impaired liver protein synthesis progress in late-stage PBC, there is a threat of reduced clotting factor synthesis[91]. Patients with PBC show lower plasma vitamin K levels in 23% of cases, which is usually accompanied by an increase in prothrombin time[92]. In patients with PBC in its end stage, portal hypertension, esophageal/gastric varices, and vitamin K deficiency increases the likelihood of massive bleeding that is difficult to stop.
Vitamin А:Vitamin A absorption requires an intact enterohepatic circulation of bile acids and formation of lipid-bile acid micellar and lamellar structures in the intestine. Significant malabsorption progression in patients with PBC, especially in those with severe cholestasis, can cause decreased intestinal vitamin A absorption accompanied by a reduction in serum retinol levels. In hepatocellular failure, the synthesis of hepatic retinol-binding protein is impaired, which also contributes to lower serum vitamin A concentrations[29]. Clinically, vitamin A deficiency is uncommon in patients with PBC. Just the same, patients with severe PBC sometimes develop insufficient dark adaptation (nyctalopia, night blindness, hemeralopia)[29]. There may be other potential manifestations of vitamin A deficiency, such as dry skin, elastosis, dermatological disorders, and impaired humoral and cellmediated immunity[29].
The developing impairment of biliary excretion processes (insufficient flow of bile acids into the duodenum) in patients with PBC in the stage of obvious cholestasis and hepatocellular failure contributes to the gradual development of fat-soluble vitamin deficiencies, which requires the use of ursodeoxycholic acid preparations, the control of plasma fat-soluble vitamin levels, and the dietary intake of foods fortified with appropriate vitamins if the latter are low. If there is deficiency of vitamin D, its active form (calcitriol) is given in combination with calcium supplements and bisphosphonates.
Changes in copper metabolism:The liver is known to play an important role in the metabolism of copper due to the hepatocyte production of ceruloplasmin-copper complexes and its excretion in bile. In health, about 80% of the copper entering the body is excreted in bile and feces.
In late-stage PBC, in which hepatocellular failure develops, copper accumulates in the liver, sometimes up to a level of 25 mg per 100 g of dry weight of liver tissue (the normal value of up to 6 mg per 100 g)[93]. At the same time, due to binding to ceruloplasmin, there are no clinical signs of copper that is toxic to humans nor is the Kayser-Fleischer ring detected.
The accumulation of copper in the body of patients with PBC leads to the activation of the coppercontaining enzyme tyrosinase. As a result, the production of melanin increases, which causes skin hyperpigmentation in these patients. With this, the body tries to excrete excess copper not only through the kidneys but also through the skin. This results in copper deposition in the epidermis.
The developing impairment of biliary excretion processes (accumulation of bile acids in hepatocytes) in patients with PBC in the stage of obvious hepatocellular failure is accompanied by abnormal copper metabolism, which requires that foods containing more than 0.5 mg of copper per 100 g of the product should be carefully incorporated into a diet, and copper utensils should not be used for cooking and storing food.
Increased metabolic rate (accelerated catabolism)
As calorie malnutrition and steatorrhea develop in PBC patients, their adaptive response leads to an increased demand of the internal organs for oxygen, which is accompanied by activation of catabolic processes, by mobilization of energy resources of adipose tissue, and by consumption of muscle protein as an energy material along with the active use of carbohydrates[54].
As glycogen stores are depleted in patients with PBC, the requirements by glucose-dependent tissues for glucose are compensated by the activation of gluconeogenesis. The latter serves as an important source for maintaining the normal glucose levels in the body and is a metabolic pathway that results in the generation of glucose from noncarbohydrate compounds. The process takes place mainly in the liver and less intensively in the renal cortex as well as in the intestinal mucosa[94]. The important precursors of glucose in gluconeogenesis are three-carbon compounds, such as lactate, pyruvate, and glycerol, which are generated by fat hydrolysis in adipocytes as well as amino acids hydrolysis of somatic (muscle) proteins. The metabolism of aromatic amino acids is known to occur predominantly in the liver, while that of branched-chain amino acids happens mainly in the muscles[43]. Patients with PBC display decreased serum concentrations of branched-chain amino acids and the increased serum level of aromatic amino acids[95-97]. In PBC patients, the increase in plasma aromatic amino acids correlates with the progression of hepatocellular failure and serves as one of its degree markers.
Unlike carbohydrates and fats, proteins and amino acids have a limited ability to be stored in the human body[94]. Amino acids are generally either used by the body as a plastic material or undergo metabolic degradation[94]. The nitrogen contained in amino acids during their degradation is converted into urea and creatinine and is excreted by the kidneys, whereas the carbon skeleton can be used for the biosynthesis of glucose (gluconeogenesis) or fatty acids or be oxidized to carbon dioxide and water to produce energy, inter alia, as ATP.
Muscles play an important role in the metabolism of amino acids, including through gluconeogenesis. Amino acids present in muscle proteins are an important source of glucose formation and metabolic energy production[94]. Glycogen and glucose deficiencies in patients with PBC enhances the catabolism of muscle (somatic) proteins to release free amino acids, many of which (primarily branched-chain amino acids) are immediately converted into pyruvate or first into oxaloacetate and then into pyruvate. The latter is converted to alanine, acquiring an amino group from other amino acids. Alanine from muscles is transported by blood to the liver, where it can be converted back into pyruvate that is used as an energy substrate or is involved in gluconeogenesis[94]. In patients with PBC, increased gluconeogenesis gradually leads to the massive breakdown and deficiency of muscle protein.
The balance between somatic protein synthesis and degradation is disturbed, which leads to the development of muscle atrophy (sarcopenia). Sarcopenia is characterized by loss of skeletal muscle mass and strength, and it is classified as secondary sarcopenia in PBC[66]. Fülsteret al[98] showed that skeletal muscle atrophy that develops in chronic diseases is also associated with low exercise tolerance. The exact mechanism contributing to sarcopenia in PBC is not clearly defined. Increased branched-chain amino acid breakdown, muscle autophagy, corticosteroids, hyperammonemia, myostatin, and physical inactivity are considered as potential contributors to sarcopenia[99,100]. The lack of amino acids and energy activates autophagy, a process in which the cell components are degraded by lysosomal enzymes. In PBC, hepatic glycogen loss, followed by accelerated gluconeogenesis, increased branchedchain amino acid catabolism, and glucocorticoid intake can result in muscle autophagy and represent an important mechanism of muscle wasting in these patients[101]. Secondary sarcopenia caused by PBC worsens quality of life and prognosis in these patients[102-104].
Osteoporosis and sarcopenia are closely related to each other and frequently coexist in patients with chronic liver diseases[105,106]. The new term “osteosarcopenia” that implies the coexistence of sarcopenia and osteoporosis has appeared[106]. Saekiet al[66] showed that the prevalence of osteosarcopenia in patients with PBC was 15.4%. Osteosarcopenia is a hazardous duet because it causes both ease of falling (due to sarcopenia) and bone vulnerability (due to osteoporosis)[106]. Osteoporosis and sarcopenia are especially problematic in postmenopausal women with PBC[66].
Protein-calorie MN and imperceptibly progressive sarcopenia gradually develop in the presence of energy deficiency, occurring in early-stage PBC in patients with clinical manifestations of cholestasis[19]. When cirrhosis develops in PBC patients, the rate of amino acid-driven gluconeogenesis significantly increases[94]. Despite the increased somatic protein degradation associated with calorie MN, the visceral pool of the protein that is synthesized in hepatocytes in PBC remains within normal limits (with minor deviations) until hepatocellular failure develops. The blood level of albumin and globulin in patients with PBC in its early stages and in the presence of severe cholestasis is within the normal range[25,26]. At the same time, the patient’s serum even in the asymptomatic stage of the disease is found to have antimitochondrial antibodies with a diagnostic titer of 1:40 and higher. As cholestasis progresses, there is an increase in the level of γ-globulins[25,26].
The developing impairment of biliary excretion processes (accumulation of bile acids in plasma and hepatocytes) in patients with PBC in the stage of obvious cholestasis results in the gradual development of protein-calorie MN following the pattern of marasmus and sarcopenia. This requires a higher protein diet (predominantly that containing branched-chain amino acids).
As PBC progresses, the resting metabolic rate and systemic thermogenesis increase due to enhanced catabolic processes[37,54]. There is a metabolic situation of resource redistribution, which is amplified as cholestasis and hepatocellular failure progress. The development of hepatocellular failure in end-stage PBC is accompanied by protein synthesizing dysfunction in hepatocytes[54]. In addition to proteincalorie MN in PBC patients during decompensated hepatocellular failure, the synthesis of urea and serum proteins decreases in the liver and the breakdown of visceral proteins increases, which causes a drastic reduction in the plasma level of circulating albumin, and there is higher urinary nitrogen excretion[107]. The continuing enhanced catabolism of somatic proteins is accompanied by the development of visceral protein deficiency, followed by edema and ascites[29]. The clinical manifestations of the impaired trophologic status in patients with end-stage PBC acquire an intermediate form of protein-calorie MN, such as marasmus-kwashiorkor. The development of PCM is facilitated by a decrease in the intestinal absorption of proteins. Portal hypertension resulting in circulatory hypoxia of the intestinal mucosa and in its increased permeability also causes a higher loss of proteins.
The developing impairment of biliary excretion processes (accumulation of bile acids in hepatocytes and plasma) in patients with PBC in the stage of obvious hepatocellular failure (a loss of 75% or more of the functioning liver cells) is accompanied by hepatocyte protein-synthetic dysfunction, which leads to visceral protein deficiency and as a consequence to the development of edema and ascites. There is a transition of the clinical form of PCM as marasmus to mixed marasmus and kwashiorkor MN. This requires a reduction in salt and fluid intake and if there are no signs of hepatic encephalopathy a higher protein diet. Nutritional support during this period should include protein modules with a predominant content of branched-chain amino acids as well as different amounts and ratios of nonessential and essential amino acids[94]. To prevent protein catabolism and to maintain nitrogen balance, it is advisable to have a meal that contains 50 g of carbohydrates before bedtime[108,109].
There is a marked improvement in the nutritional status of PBC patients at the stage of development of cirrhosis and resistant ascites after successful treatment of the latter, which emphasizes the importance of nutritional support in these patients[110].
Hepatic encephalopathy:In end-stage PBC, progressive hepatocellular failure, portal hypertension, and portosystemic shunting lead to hepatic encephalopathy (HE)[111,112]. HE is considered to mean potentially reversible neuropsychiatric disorders, the development of which is based on detoxifying dysfunction of liver and portal blood shunting, developing in the presence of severe liver injuries[111,113]. HE is a classic symptom of advanced hepatocellular failure[111,112]. In prognostic terms, the encephalopathy associated with progressive hepatocyte death becomes a formidable and almost always fatal complication of PBC. The prevalence of minimal HE among patients with liver cirrhosis ranges from 30% to 84%[114].
There is a metabolic theory of HE, which is based on the reversibility of its main symptoms in very extensive cerebral disorders[115]. In PBC, one can identify two factors that determine the relationship between the liver and the nervous system and play a role in the pathogenesis of HE[113]: The ability of the liver to detoxify neurotoxic poisons (ammonia, mercaptan, skatole, indole, phenols,etc) produced in the intestine by the digestion of food ingredients and as a result of vital microbial activity[116,117].
Cerebral metabolism strongly depends on the maintenance of the normal glycemic level that is appreciably determined by the storage of glycogen in the liver and the rate of glycolysis between meals. As mentioned above, the glycogen stores are depleted in PBC. A decrease in the intensity of oxygen and glucose metabolism in PBC is accompanied by reductions in energy production and neuronal activity, which contributes to the development of signs of HE[115]. Positron emission tomography shows that in HE there is a strong correlation between the reduction in cerebral blood flow (in the frontal and parietal lobes of the cerebral cortex), which is accompanied by decreased glucose metabolism and the results of neuropsychological tests[113].
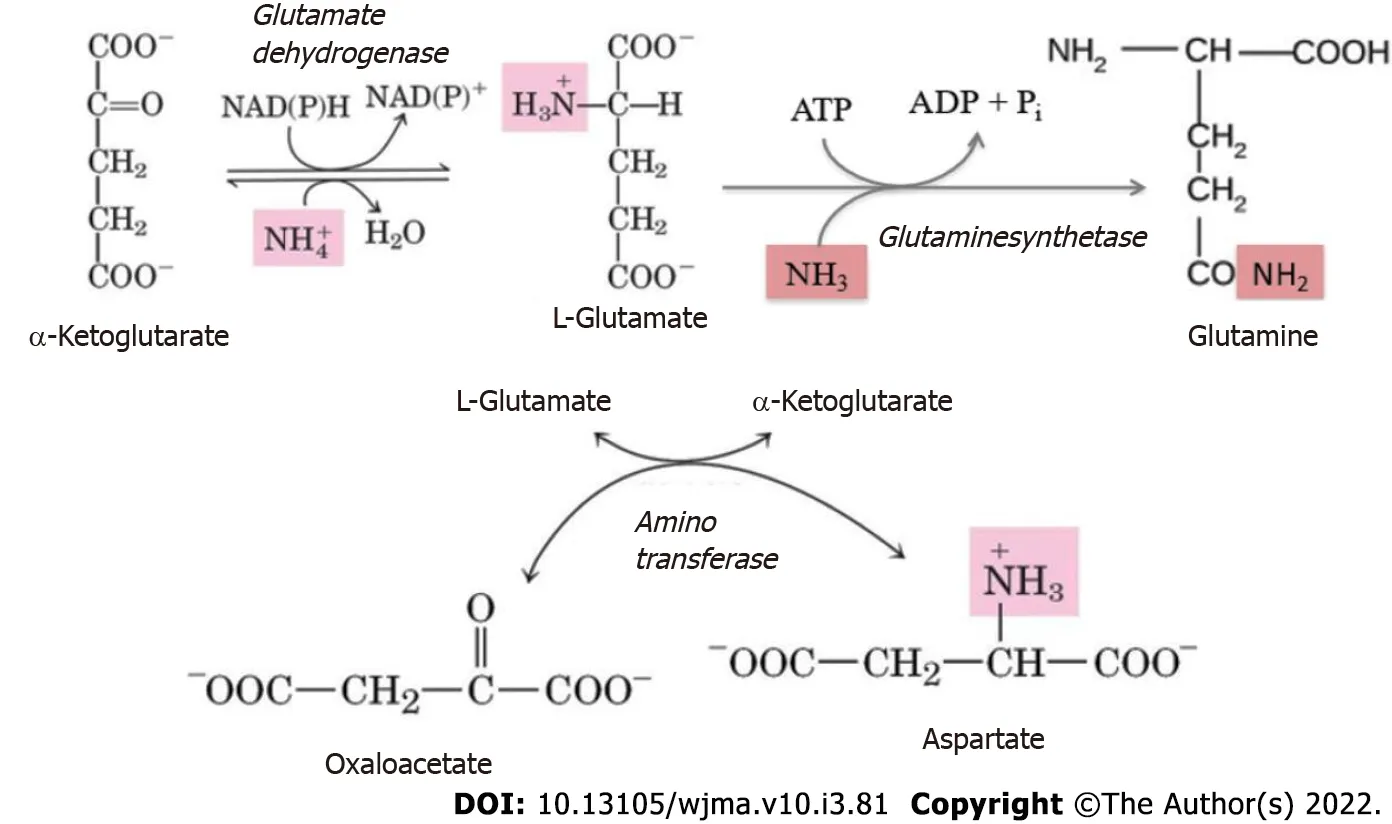
Figure 4 Use of α-ketoglutarate, glutamate, oxaloacetate, and aspartate to detoxify ammonia.
The basis for the pathogenesis of HE is hepatocellular failure, accompanied by a decrease in the hepatic clearance of neurotoxic poisons produced in the intestine by the digestion of food ingredients and as a result of vital microbial activity, portosystemic shunting, and amino acid metabolic disturbance that gives rise to false neurotransmitters[115].
Protein and amino acid degradation results in the formation of amine nitrogen that, unlike the hydrocarbon portion of amino acids, is unsuitable for energy production[94]. Therefore, the amino groups that cannot be reused, for example, in transamination reactions, are converted to ammonia. The latter in the cells is produced by the deamination of amino acids, nucleotides, and biogenic amines. Ammonia is a toxic substance and its blood concentration in health does not exceed 50 μmol/L. In health, about 7% of the ammonia formed in the body passes through the tissue of the brain without causing any changes in its functions[111]. The fundamental reaction of ammonia neutralization, which takes place in all tissues, is the binding of NH3to glutamate to form glutamine (Figure 4). Its major tissue suppliers of glutamine are muscles, brain, and liver.
In addition to ammonia formed in tissues, significant amounts of NH3are generated in the intestine by the bacterial microflora and as a result of food protein hydrolysis. The intestinal absorption of ammonia can cause its significant supply to the liver. This occurs with intake of higher protein foods, incomplete bowel evacuation, alkalization of intestinal contents, overgrowth of opportunistic pathogens, and bleeding esophageal/gastric varices with the development of portal hypertension[112]. The concentration of toxic products, primarily ammonia, as well as skatole, indole, and phenols, thereof may increase in the intestine. In health, these substances enter the portal venous system and undergo the ornithine cycle in the liver. Through deamination, transamination, and decarboxylation reactions, they are converted to urea, a product that is relatively harmless for the body[94]. Urea is the major end product of nitrogen metabolism (85% of all nitrogen is excreted from the body with urea). Urea in the human body is synthesized only in the liver[91].
Neuronal dysfunction results from elevated neurotoxic ammonia levels in the blood in hyperammonemia[110]. The latter is observed in patients with PBC in the cirrhosis developmental stage and is caused by the increased intestinal absorption of ammonia, its impaired hepatocyte detoxification (reduced urea cycle enzyme activity), and lower ammonia binding in hypotrophic skeletal muscles (decreased glutamine synthetase activity)[112,118]. The disturbed hepatic blood flow is of great importance in the development of HE. The development of cirrhosis in end-stage PBC causes blood to shunt either inside the liver itself (portal hepatic venous anastomoses that function as intrahepatic shunts form around the lobules) or blood to flow from the portal vein into the general circulation, bypassing the liver through natural collaterals[115]. Portosystemic shunting and collateral blood flow pathways cause blood flowing from the intestine to enter the general circulation, bypassing the liver. The toxic substances and primarily ammonia, which are contained in the blood portal system, enter the general bloodstream non-neutralized.
Hyperammonemia in patients with PBC triggers compensatory mechanisms of the metabolism and clearance of ammonia, by activating the processes of its neutralization in skeletal muscles and neurons[119]. The elevated blood level of ammonia results in its increased penetration through the blood-brain barrier into the brain, which has an adverse effect on astrocytes. Ammonia detoxification in the astrocytes is affected by glutamate synthetase, leading to the binding of ammonia to glutamate to yield glutamine (Figure 4)[120].

Figure 5 Synthesis of false neurotransmitters in hyperammonemia.
Excess ammonia in the muscle tissues can also be inactivated due to its interaction with both glutamate and aspartate to synthesize glutamine (Figure 4)[113,121]. When ammonia is excessive, there is a depletion of glutamate and aspartate stores (with simultaneous accumulation of glutamine). The larger amount of glutamine produced is released into the bloodstream in exchange for branched-chain amino acids[122].
In PBC, hyperammonemia thus requires increased production of glutamate and aspartate from αketoglutarate and oxaloacetate. This causes a portion of α-ketoglutarate and oxaloacetate to leak away from the tricarboxylic acid cycle, which is accompanied by decreased ATP synthesis. Since the neurons are especially sensitive to decreased energy production, this fact plays a role in the mechanism for the development of clinical signs of HE and causes enhanced energy deficiency in patients with PBC.
The muscles are also sensitive to decreased ATP energy production. Thus, to increase the levels of αketoglutarate and oxaloacetate in muscles, which are needed for the Krebs cycle, and to maintain a sufficient glutamate level, accelerated branched-chain amino acid catabolism occurs in PBC patients with hyperammonemia. This results in the insufficient synthesis of muscle proteins and their depletion[123,124]. Hyperammonemia is associated with HE, enhanced branched-chain amino acid catabolism, and sarcopenia[125]. Sarcopenia exacerbates HE, which in turn aggravates a decline in food intake and the development of MN. There is a vicious circle that is difficult to break.
There is evidence that hyperammonemia affects the saturation center in the hypothalamus and suppresses appetite, which can also increase protein-calorie MN in patients with PBC[111,113].
Along with hyperammonemia, a disturbance in the synthesis and metabolism of the major neurotransmitters derived from the aromatic amino acids tyrosine and phenylalanine plays an important role in the pathogenesis of HE in patients with PBC at the stage of development of hepatocellular failure and portosystemic shunting[115]. ter Borget al[39] found elevated concentrations of the aromatic amino acids, tyrosine and phenylalanine, and decreased blood concentrations of the branchedchain amino acids, valine, isoleucine, and leucine, in patients with PBC both at the stage of development of cirrhosis and without cirrhosis[39]. Increased entry of aromatic amino acids into the blood (due to their impaired catabolism in the liver) inhibits the enzyme systems involved in the conversion of aromatic amino acids to catecholamines, which reduces the biosynthesis of dopamine and norepinephrine and increases the synthesis of serotonin from tryptophan. Entering the brainviathe blood-brain barrier, tyrosine and phenylalanine are involved in the synthesis of false neurotransmitters, such as β-phenylethanolamine and octopamine (Figure 5).
Tyramine is formed from the amino acid tyrosine under the action of bacterial decarboxylases in the intestine. The former is a physiologically active and toxic substance. It easily enters the general circulation and penetrating the blood-brain barrier affects excitatory and inhibitor processes in the nervous system when patients with PBC undergo portosystemic shunting. Along with hyperammonemia, false neurotransmitters (tyramine) inhibit neuronal function and enhance HE progression[39]. Cognitive impairment, forgetfulness, feeling sleepy during meals and snacks, and having difficulties in cooking food when HE develops in late-stage PBC are significant challenges faced by patients in this group[126]. Subsequently, MN itself becomes an independent predictor of mortality in patients with PBC.
The developing impairment of biliary excretion processes (accumulation of bile acids in hepatocytes) in patients with PBC causes obvious hepatocellular failure, which is accompanied by hepatocyte detoxifying dysfunction, hyperammonemia, and the formation of false neurotransmitters with the development of HE. This requires a strict dietary protein restriction, branched-chain amino acid diets (containing a minimum amount of aromatic amino acids), the prescription of antibiotics (the effect of which is based on their effect on microorganisms that produce nitrogenous compounds in the gastrointestinal tract), aimed at a change in the ratio of neurotransmitters, a decrease in the formation and absorption of ammonia and other toxins formed in the intestine, and an increase in the elimination of ammonia. Strict vegetarian diets (vegetable protein up to 120 g/d) and dairy proteins are generally well tolerated (presumably due to the low aromatic amino acid levels). During this period, patients with PBC usually need to be placed on a waiting list to undergo liver transplant surgery. The European Society for Clinical Nutrition and Metabolism guidelines are used in clinical practice to meet the energy and protein requirements of patients with MN and weight loss in surgical or intensive care units.
Poor nutritional status has serious implications for postoperative complications among candidates for liver transplantation, as it is an important predictor of mortality and postoperative complications in patients with PBC.
CONCLUSlON
PBC is a chronic, slowly progressive disease of the liver and biliary tract, which results in a change in the trophological status of these patients. The causes of MN in PBC are complex and multifactorial since the liver is involved in many metabolic processes of the body. But the leading role in the development of MN in patients with PBC is played by a disturbance in biliary excretion processes, and as a consequence there are changes in the metabolism of macronutrients and micronutrients. Trophological insufficiency develops gradually and imperceptibly as cholestasis progresses with the insufficient entry of bile acids in the duodenum with their simultaneous deposition in hepatocytes and entrance into the general circulation. It is precisely these changes that trigger the development of calorie (energy) MN, even in the asymptomatic and early stages of the disease. Compensatory mechanisms for obtaining energy from fatty acids and amino acids of somatic proteins are turned on with time, which is accompanied by protein-calorie (as marasmus) MN with a slowly progressive weight loss. Lipid metabolism disorders also develop. There is an increased synthesis of cholesterol and phospholipids to neutralize the detergent effect of excess plasma bile acids. Insufficient intestinal entry of bile acids contributes to the development of steatorrhea and fat-soluble vitamin deficiencies in these patients. Hence, an increase in PCM occurs and vitamin and mineral deficiencies gradually progress. The latter gives rise to osteoporosis and osteosarcopenia. Prolonged exposure of hepatocytes to excessive bile acid concentrations leads to liver fibrosis and cirrhosis, portal hypertension, and portosystemic shunting to impair protein synthesizing and detoxifying functions of the liver. Occurring visceral protein deficiency results in edema, ascites, and increased PCM with a transition to the mixed form of marasmus and kwashiorkor. Developing hyperammonemia and resulting false neurotransmitters lead to changes in the central nervous system, and HE develops. MN progresses as the severity of the disease progresses. All this makes the correction of MN especially difficult in patients with PBC. Thus, assessment of nutritional status and control of MN are of great importance for improving treatment outcomes in these patients. The presented mechanisms of trophological changes in PBC should assist in timely recognition of MN and in correctly selecting a nutrition support regimen for these patients at different stages of disease development along with symptomatic therapy.
ACKNOWLEDGEMENTS
The authors are grateful to Tatiana Igorevna Karlovich and Alexander Igorevich Burmistrov for discussions and technical assistance in preparing the review for publication.
FOOTNOTES
Author contributions:Reshetnyak VI and Maev IV have equally contributed to the conception and design of the study, literature review and analysis, drafting and critical revision and editing, and final approval of the final version.
Conflict-of-interest statement:All the authors declare that they have no conflict of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Russia
ORClD number:Vasiliy Ivanovich Reshetnyak 0000-0003-3614-5052; Igor Veniaminovich Мaev 0000-0001-6114-564X.
S-Editor:Liu JH
L-Editor:Filipodia
P-Editor:Liu JH
 World Journal of Meta-Analysis2022年3期
World Journal of Meta-Analysis2022年3期
- World Journal of Meta-Analysis的其它文章
- Responses to disrupted operative care during the coronavirus pandemic at a Caribbean hospital
- Viral hepatitis: A narrative review of hepatitis A-E
- Rare post-endoscopic retrograde cholangiopancreatography complications: Can we avoid them?
- Review with meta-analysis relating North American, European and Japanese snus or smokeless tobacco use to major smoking-related diseases
- Evidence analysis on the utilization of platelet-rich plasma as an adjuvant in the repair of rotator cuff tears
- ls cellular therapy beneficial in management of rotator cuff tears? Meta-analysis of comparative clinical studies