
基于低温热释放的烷基接枝相变偶氮苯材料
2022-08-22 10:59冯奕钰方文宇
高等学校化学学报 2022年8期
高 健,冯奕钰,2,方文宇,王 慧,葛 婧,封 伟,2
(1.天津大学材料科学与工程学院,天津 300354;2.天津大学先进陶瓷与加工技术教育部重点实验室,天津 300072)
太阳能因储量丰富、分布广、可持续性强而被认为是最具潜力的可再生能源[1~3]. 利用分子光开关将太阳能存储在化学键能中是最具吸引力的太阳能转换和储存方式[4,5]. 由于偶氮苯及其衍生物具有良好的光响应性、可逆的顺反异构化和结构可调控等优点,被认为是最具潜力的太阳能热电池[6]. 然而,偶氮苯能量密度低、储能半衰期短、异构化速率慢,难以满足未来的需求[7]. 目前解决上述问题的主要方法是在偶氮苯体系中引入分子间氢键和配位键等,并将其接枝到碳基[8,9]或聚合物基[10,11]模板中. Feng 等[12~14]通过优化分子间/内相互作用,获得了一系列高能量密度(720 J/g)和长储能半衰期(160 h)的偶氮苯/石墨烯复合材料. Venkataraman 等[15]通过将偶氮苯单元接枝到聚合物上制备了一种聚合物模板化的偶氮苯材料,该材料同样表现出高能量密度(698 J/g).
虽然模板的引入极大地提高了偶氮苯的能量密度,但其密实的结构、复杂的制备过程以及较慢的异构化速率限制了偶氮苯在低温环境中的应用[16]. 偶氮苯具有光诱导可逆固-液相变的特性,即在异构化后通过相变温度的变化可由固体变为液体[17,18]. 目前研究中,此类偶氮苯多用于与织物进行复合,制备可穿戴的热管理器件[19].偶氮苯分子中存储着两种能量,异构化焓(ΔHiso,J/g)和相变焓(ΔHc,J/g),它们可以通过可见光激发同时释放出来,这为实现低温放热提供了机会[20,21]. 因此,合成具有合适熔融温度(Tm,℃)或结晶温度(Tc,℃)的相变偶氮苯是解决低温放热问题的关键. Wu等[22]总结了单取代和双取代偶氮苯的熔点,大部分偶氮苯结晶能力强,熔点在20~200 ℃,这意味着偶氮苯在低温下的热释放仍然是一个挑战.
本文合成了两种不同取代基的烷基接枝相变偶氮苯——4-正戊基偶氮苯(Azo5)和2′-甲基-4-正戊基偶氮苯(AzoM5). 通过控制苯环上是否有甲基取代基,有效地降低异构化前后的Tm和Tc. 相变偶氮苯由于光热能和环境热的同时存储与释放,具有较高的能量密度. 最后,在-10 ℃的环境下实现了储热材料的放热可视化.
1 实验部分
1.1 试剂与仪器
4-正戊基苯胺(纯度97%)和亚硝基苯(纯度98%),日本TCI公司;2-亚硝基甲苯,纯度97%,美国默克公司;石油醚(沸程60~90 ℃)、甲苯和乙酸乙酯,分析纯,天津市江天化工技术股份有限公司;冰乙酸,分析纯,天津市元立化工有限公司.
Avance ⅢHD 400 MHz型液体核磁共振波谱仪(1H NMR),德国布鲁克公司,以氘代二甲基亚砜为溶剂;UV-3600 型紫外-可见光谱仪(UV-Vis),日本岛津有限公司;IN10 型傅里叶变换红外光谱仪(FTIR),KBr压片,美国赛默飞公司;DSC214型差示扫描量热仪(DSC),德国耐驰公司;SLTD2-500PS型半导体恒温试验台,天津精易工贸有限公司;Tix 640型红外热成像仪,美国福禄克公司.
1.2 Azo5和AzoM5的制备与表征
参照文献[23]报道的Mills法制备Azo5. 合成路线如Scheme 1所示. 将0.54 g(5 mmol)亚硝基苯和50 mL 甲苯进行超声溶解后加入到100 mL 三口烧瓶中,加入0.82 g(5 mmol)4-正戊基苯胺和1.14 mL(20 mmol)冰乙酸,搅拌,于60 ℃下回流反应24 h;将得到的粗产物通过柱层析法提纯,洗脱剂为石油醚-乙酸乙酯(体积比20∶1),最终得到Azo5液体1.12 g.
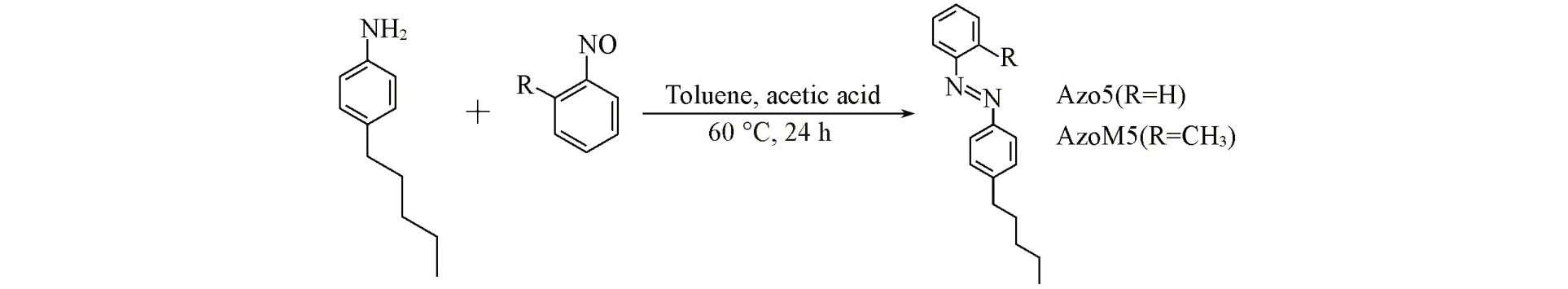
Scheme 1 Synthesis of Azo5 and AzoM5
以0.61 g(5 mmol)2-亚硝基甲苯代替0.54 g(5 mmol)亚硝基苯,采用与合成Azo5相同的方法制备AzoM5,得到AzoM5液体1.14 g.
利用UV-Vis光谱研究样品的异构化性能:在室温(20 ℃)下,将5 μL样品置于干净玻璃片上,用紫外光(365 nm,150 mW/cm2)照射,间隔一定时间后,取微量样品溶于乙酸乙酯中,进行UV-Vis 光谱测试,测试范围为250~600 nm. 黑暗自然回复和可见光(420 nm,80 mW/cm2)照射回复的测试过程与上述类似;用DSC 测试样品的热性能,反式异构体采用以下过程:在40 ℃平衡,以2 ℃/min 的速率降温至-20 ℃,保温1 min;以2 ℃/min 的速率加热至40 ℃;顺式异构体采用以下过程:在20 ℃平衡,以2 ℃/min的速率降温至-100 ℃,保温1 min;以2 ℃/min的速率加热至150 ℃;使用红外热成像仪(IR)分别测试-10 ℃环境下Azo5反式和顺式异构体放热引起的温度变化,其中紫外光和可见光的波长、功率与异构化性能测试中一致.
2 结果与讨论
2.1 光致相变机理
偶氮苯及其衍生物因异构化前后极性和体积等发生变化,通常导致其反式异构体(Trans)比顺式异构体(Cis)具有更高的Tc和Tm[24]. 由Scheme 2可见,当环境温度处于反式异构体的Tc和顺式异构体的Tc之间时,反式异构体吸收光热能变为顺式异构体,同时自发地吸收环境热发生固-液转变,实现了光热能和环境热的共同收集. 顺式异构体在外界刺激下(热或可见光)发生异构化回复,与此同时发生相变,由顺式液体变为反式固体,将储存的能量以ΔHiso和ΔHc的形式释放. 相比于非相变偶氮苯只有异构化焓的释放,光控固-液相变的发生使相变偶氮苯具有更大的能量密度.

Scheme 2 Schematic diagram of the simultaneous photo⁃induced solid⁃to⁃liquid and trans⁃to⁃cis isomerisation transitions
2.2 结构表征
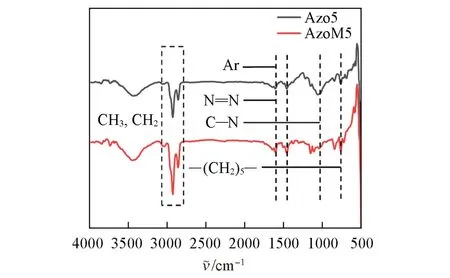
Fig.1 FTIR spectra of Azo5 and AzoM5
图1 为Azo5 和AzoM5 的FTIR 谱图. 由图1 可知,Azo5 和AzoM5 的甲基(CH3)和亚甲基(CH2)的伸缩振动峰位于2925和2858 cm-1处;苯环骨架结构(Ar)的吸收峰位于1603 cm-1处;氮氮双键(N=N)的吸收峰位于1458 cm-1处;碳氮单键(C—N)的吸收峰位于1109 cm-1处;长烷基链[—(CH2)n—]的水平摇摆吸收峰位于723 cm-1处,初步证明成功合成烷基接枝的偶氮苯. 图2(A)为Azo5 的1H NMR谱图. 图中Azo5 的氢原子化学位移分别出现在δ7.91~7.83, 7.86~7.79, 7.64~7.51, 7.45~7.39,2.67,1.63,1.30,0.91~0.79处,积分面积比为2∶2∶3∶2∶2∶2∶4∶3,与理论比值一致,证明Azo5 被成功合成. 图2(B)为AzoM5 的1H NMR 谱图. 图中AzoM5 的氢原子化学位移分别出现在δ7.85~7.78,7.55,7.48~7.37,7.36~7.27,2.67,1.62,1.40~1.14,0.91~0.78 处,积分面积比为2∶1∶4∶1∶5∶2∶4∶3,与理论比值一致,证明成功合成了AzoM5.
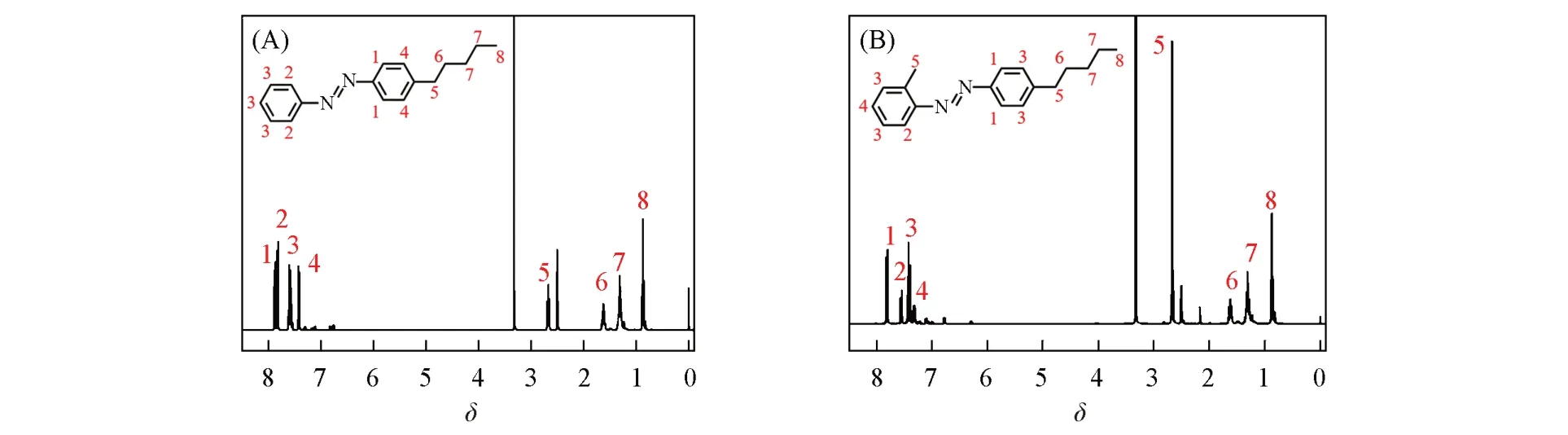
Fig.2 1H NMR spectra of Azo5(A)and AzoM5(B)
2.3 异构化性能
偶氮苯的异构化性能是能量存储与释放的基础,通过UV-Vis光谱可以充分地了解偶氮苯的异构化性能. 由图3(A)和(B)可见,Azo5 和AzoM5 在紫外光照射下可以实现由反式到顺式的异构化转变,330 nm处的吸收峰强度显著下降,440 nm处的吸收峰强度略有上升. Azo5和AzoM5从反式转变为顺式异构体的照射时间不同,分别为42和30 min,说明苯环上甲基的存在会影响偶氮苯的异构化速率. 由于甲基的存在破坏了π-π堆积,使分子间的空间位阻降低,有助于其异构化转变,这一现象在异构化回复中也得到证明[25]. 由图3(C)和(D)可见,Azo5和AzoM5在黑暗条件下自发地发生由顺式到反式的异构化回复,自发回复时间分别为168和144 h,有助于能量的长效存储. 由图3(E)和(F)可见,在可见光照射下,Azo5 和AzoM5 由顺式到反式的异构化回复能够被加速,回复时间远远小于黑暗条件下的,这有助于实现可控放热.

Fig.3 UV⁃Vis absorption spectra of Azo5(A,C,E)and AzoM5(B,D,F)corresponding to isomerization upon irradiation with UV light(365 nm, A, B), in the dark(C, D) or with visible light(420 nm,E,F)in different times
除异构化速率外,异构化程度和循环稳定性也是评价偶氮苯储能性能的重要指标. 偶氮苯的储能容量随异构化程度的增加而增大,而循环稳定性决定了其使用寿命. 异构化前后,因为氢原子的化学环境发生变化,从而导致其在1H NMR中的化学位移会发生变化,所以可以利用1H NMR结果计算其异构化程度. 根据图4(A)和(B)中异构化前后1H NMR谱中δ1.50和1.60处的峰面积比,确定了偶氮苯的异构化程度. 经计算,Azo5 和AzoM5 在溶剂辅助充热下达到稳态时的异构化程度分别为77%和82%. 通过UV-Vis与1H NMR联用,计算出Azo5和AzoM5在无溶剂辅助充热下达到稳态时的异构化程度分别为61%和67%,与之前的报道值(60%~70%)相当[26,27]. 溶剂的加入会削弱分子间π-π堆积作用和范德华力,提高异构化程度;而AzoM5中甲基的存在增加了分子间的距离,也会削弱分子间π-π堆积作用和范德华力,从而使AzoM5 比Azo5 具有更高的异构化程度. 为验证其循环稳定性,将Azo5 和AzoM5分别用紫外光和可见光交替照射,并记录其在330 nm处吸光度的变化. 由图5(A)和(B)可见,Azo5和AzoM5在10次循环中表现出良好的循环稳定性,吸光度几乎没有衰减(<1%),这有利于光热存储材料的长期循环利用.

Fig.4 1H NMR spectra of trans⁃and cis⁃Azo5(A)and AzoM5(B)

Fig.5 Cycling stability of Azo5(A)and AzoM5(B)under alternating UV light and visible light irradiation
2.4 储热与放热性能
利用DSC测试了Azo5和AzoM5反式和顺式异构体的储热与放热性能. 由图6(A)和(B)可见,在反式状态下,AzoM5比Azo5具有更低的Tm和Tc,这是因为苯环上甲基的存在破坏了对称性,导致AzoM5的结晶能力更弱[28]. 由图6(C)和(D)可见,在顺式状态下,Azo5和AzoM5在降温过程中都不具有结晶行为,但是在升温过程中Azo5出现了冷结晶现象,这是因为其结晶能力弱,降温过程中链段来不及运动,只有通过热刺激才能激活链段的运动,从而实现晶体的生长[29]. 由于Azo5和AzoM5的顺式异构体都不具有结晶行为,理论上其在反式异构体的Tc(-1 ℃,-15 ℃)以下均可以实现光控的固-液转变.
能量密度是设计高效太阳能热电池的最关键因素,它直接反映了太阳能热电池的储能能力. 相变偶氮苯的总能量密度(△Htotal,J/g)分为两部分,一部分是反式异构体的△Hc(J/g),另一部分是△Hiso(J/g). 其中△Hc和△Hiso可以分别由反式异构体DSC曲线的结晶放热峰和顺式异构体DSC曲线的异构化放热峰的积分面积得到. 根据下式计算△Htotal:

Azo5和AzoM5的△Htotal分别为216和218 J/g,因结晶焓的引入使其能量密度显著提高. 此外,△Hc和△Hiso的同时释放有利于实现环境热和光热能的集中利用,对于实现低温放热具有很大的价值.
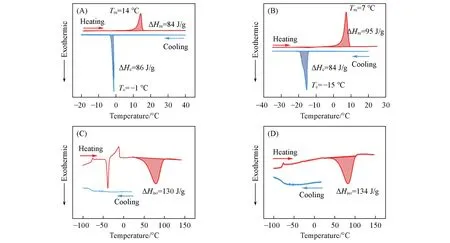
Fig.6 DSC diagrams of trans⁃Azo5(A)and trans⁃AzoM5(B),cis⁃Azo5(C)and cis⁃AzoM5(D)

Fig.7 Morphology of Azo5 during exothermic processes(A), change in temperature of Azo5 with time(B) and IR thermal⁃imaging of cis⁃Azo5(top) and trans⁃Azo5(down) under visible light irradiation at-10 ℃(C)
以Azo5为例,研究了其对环境热和光热能的收集与释放. 由图7(A)可见,整个能量循环过程由4个步骤组成:(1)充热. 在紫外光照射下由橙色液体变为红色液体.(2)降温. 降温至Tc以下.(3)放热. 在可见光照射下,由红色液体变为橙色固体.(4)升温. 吸收环境热进行蓄热,由橙色固体变为橙色液体. 通过IR摄像机跟踪Azo5放热过程中的温度变化[图7(B)和(C)]. 在可见光照射下,Azo5的反式和顺式异构体温度均有所上升,但顺式异构体具有更高的升温速率,并且在73 s 时达到最高温度(-2.7 ℃),比起始温度高6 ℃,这归因于结晶焓和异构化焓的同步释放. 在73 s后,顺式异构体的温度逐渐下降,这是因为热量的释放不足以弥补向低温环境扩散的热量. 反式异构体在40 s后温度趋于稳定,达到-6.9 ℃,比起始温度高1.7 ℃,这归因于可见光的光热效应. 在240 s 后停止可见光照射,Azo5的反式和顺式异构体的温度均迅速下降回起始温度. 通过对比可知,去除光热效应的影响,顺式异构体通过结晶焓和异构化焓的同步释放可以实现4.3 ℃的温度提升.
3 结论
合成了两种不同取代基的烷基接枝相变偶氮苯(Azo5,AzoM5)并研究了低温下其对环境热与光热能的利用性能. UV-Vis光谱分析结果表明,两者皆具有快速的光致异构化和良好的循环稳定性,并且可见光照射可以加速异构化回复,为可控热释放奠定基础. DSC分析结果表明,两者反式异构体皆具有较低的Tc,并且顺式异构体不具有结晶行为,可以实现低温下的光致固-液转变. Azo5展示了-10 ℃环境下的放热性能,在可见光照射下实现了6 ℃的温度提升. 结果表明,该材料不仅可以同时存储光热能和低温环境热量,还可以展现出可控的热量释放,未来有望与织物复合制备低温可穿戴器件.
猜你喜欢
哈尔滨理工大学学报(2022年1期)2022-05-10
浙江化工(2022年4期)2022-05-07
化学研究(2022年1期)2022-02-27
临床与实验病理学杂志(2021年7期)2021-09-06
石油石化绿色低碳(2020年2期)2020-12-31
探索科学(学术版)(2019年8期)2020-01-17
化工管理(2020年1期)2020-01-16
物理学报(2019年12期)2019-06-29
健康博览(2018年7期)2018-01-03
中国粮油学报(2014年8期)2014-02-06
