
Evidence-based pathogenesis and treatment of ulcerative colitis: A causal role for colonic epithelial hydrogen peroxide
2022-08-18 03:16JayPravda
World Journal of Gastroenterology 2022年31期
Jay Pravda
Abstract In this comprehensive evidence-based analysis of ulcerative colitis (UC), a causal role is identified for colonic epithelial hydrogen peroxide (H2O2) in both the pathogenesis and relapse of this debilitating inflammatory bowel disease. Studies have shown that H2O2 production is significantly increased in the non-inflamed colonic epithelium of individuals with UC. H2O2 is a powerful neutrophilic chemo-tactic agent that can diffuse through colonic epithelial cell membranes creating an interstitial chemotactic molecular “trail” that attracts adjacent intra-vascular neutrophils into the colonic epithelium leading to mucosal inflammation and UC.A novel therapy aimed at removing the inappropriate H2O2 mediated chemotactic signal has been highly effective in achieving complete histologic resolution of colitis in patients experiencing refractory disease with at least one (biopsy-proven)histologic remission lasting 14 years to date. The evidence implies that therapeutic intervention to prevent the re-establishment of a pathologic H2O2 mediated chemotactic signaling gradient will indefinitely preclude neutrophilic migration into the colonic epithelium constituting a functional cure for this disease.Cumulative data indicate that individuals with UC have normal immune systems and current treatment guidelines calling for the suppression of the immune response based on the belief that UC is caused by an underlying immune dysfunction are not supported by the evidence and may cause serious adverse effects.It is the aim of this paper to present experimental and clinical evidence that identifies H2O2 produced by the colonic epithelium as the causal agent in the pathogenesis of UC. A detailed explanation of a novel therapeutic intervention to normalize colonic H2O2, its rationale, components, and formulation is also provided.
Key Words: Ulcerative colitis; Pathogenesis, Treatment; Hydrogen peroxide
lNTRODUCTlON
Treating ulcerative colitis (UC) has never been easy. The natural history of UC is one of worsening and progressive disease, and no currently available approved medication can cure the life-long repeating episodes of rectal bleeding, diarrhea, and abdominal pain that are experienced by individuals suffering from this illness[1]. The difficulty in choosing from currently available non-curative therapies was underscored by a recent study, which concluded that the majority of clinical guidelines for the treatment of UC are based on low or very low-quality evidence[2]. Thus, we are left with therapies that cannot cure and have a disappointing track record when it comes to treatment.
The distress engendered by the lack of effective treatment is universal with the majority of UC patients in a 10-country global survey reporting poor disease control, mental exhaustion, and adverse impact on quality of life[3]. This is consistent with other multi-country studies reporting that UC was not controlled in over 87% of participants[4]. The real-world effects of non-curative low-quality therapy are evident by the high degree of medical treatment failure that is responsible for up to a 30% colectomy rate in patients with this illness[5]. With a dismal 40% one-year clinical remission rate for current drugs that alter the immune response, and similar upcoming drugs no more effective, there is no reason to believe that any treatment focused on modifying the immune response will improve current patient outcomes[6]. We are thus left to conclude that this class of therapeutics has reached the limit of clinical effectiveness, and any hope for effective therapy or a cure can only arrive with a fundamentally new approach in our understanding and treatment of this disease.
Almost all treatments for UC consist of agents that modify, alter, or suppress the immune response[7,8]. This is based on the belief that an underlying immune abnormality is the cause of this condition. But is this assumption evidence-based? Unfortunately not, despite extensive research conducted since the mid-20thcentury, no evidence of a causal antecedent immune vulnerability has been uncovered in individuals with UC or their first-degree relatives[9]. Additionally, studies in UC patients have revealed normal immune responses when compared to healthy controls[10,11]. Thus, the evidence indicates that an immune abnormality is not the cause of UC, and treatment directed against the immune response cannot bring about a cure, restore healthy colonic functionality, or a normal quality of life.
Faced with these facts, we must consider that the immune response in UC is an accompanying effect of a separate underlying phenomenon that has a causal role in the development of this disease. In other words, the immune system is doing what it’s programed to do given the stimulus it is subjected to. But if there’s nothing wrong with the immune system then what stimulus could cause inflammation of the colon leading to UC and how can we treat it? The next section describes a novel evidence-based pathogenesis that provides answers to these questions.
UC: AN EVlDENCE-BASED PATHOGENESlS
A causal role for colonic epithelial cell hydrogen peroxide in the pathogenesis of UC
In order to understand the pathogenesis of UC and develop an effective treatment, we need to answer several questions. Starting with what we can see, we must explain why the inflammation typically begins in the rectum and advances contiguously to more proximal regions of the colon without sparing intervening mucosa. We also need to identify the molecular mechanism that initiates the inflammation in the first place. In other words, how the inflammation begins. This raises the question of what causes this mechanism to initially appear and reappear over and over again after (apparently) successful treatment leading to life-long relapse. Finally, we need to derive the genetic predisposition that makes this all possible. Understanding the overlapping lineal sequence of events leading up to UC and the mechanism of relapse is crucial for effective therapeutic intervention and long-term remission so as to permanently alter the natural history of disease. Stated differently, we will start with what we can see(the inflammation) and work our way upstream until we arrive at the inception of disease, which originates from the interaction of a shared genetic predisposition with exposomal elements giving rise to a final common pathway that must be present among all individuals with UC; at all times basing our conclusions on the known experimental evidence.
Neutrophils are the first responders into the colonic epithelium in UC with the formation of neutrophilic cryptitis and neutrophilic crypt abscesses, which are hallmarks of active inflammation[12-14]. This typically begins in the rectum causing mucosal inflammation, which advances proximally and contiguously (without skipping). Once in contact with bacteria in the rectal epithelium, neutrophils are activated to release large amounts of hydrogen peroxide (H2O2). Studies have shown that a single neutrophil can produce enough H2O2to diffuse into and oxidize nearly all the hemoglobin contained in ten intact surrounding red blood cells[15].
H2O2is a cell-membrane permeable, highly potent neutrophilic chemotactic factor that attracts neutrophils into the colonic epithelium[16]. Studies have demonstrated that neutrophils can respond to and migrate towards an H2O2concentration variation of 100 picomolar, which is a difference of approximately five molecules of H2O2between the leading and trailing halves of the neutrophil[17]. H2O2is also a powerful oxidizing agent that disintegrates tight junctional proteins[18-21]. This leads to increased paracellular permeability and decreased epithelial resistance, which is characteristically observed in UC[22-25].
The resulting H2O2mediated increase in paracellular permeability facilitates antigenic translocation across the colonic epithelium while simultaneously creating an H2O2chemotactic gradient, both of which act cooperatively to attract other neutrophils into the advancing proximal edge of the inflammatory field thereby extending colonic inflammation from the rectum, in a contiguous fashion, to more proximal regions of the colon. The inflammation only halting upon encountering sufficient circumferential epithelial reductive capacity to neutralize the advancing wave of neutrophil released H2O2,resulting in a sharp demarcation between healthy and diseased tissue. This redox tug-of-war between epithelial reductive capacity and neutrophilic H2O2explains the characteristic proximal migratory behavior of colonic mucosal inflammation in UC. This interpretation is supported by studies showing that neutrophil accumulation within epithelial crypts and in the intestinal mucosa directly correlates with clinical disease activity and epithelial injury in individuals with UC[26]. Stated differently,neutrophils in the crypts of Lieberkühn secrete large amounts of H2O2that attracts other neutrophils into the epithelium. Continuous secretion of H2O2by neutrophils overwhelms epithelial reductive(antioxidant) capacity causing additional neutrophils to enter the inflammatory field. This advances the inflammation in a proximal direction until sufficient epithelial reductive (antioxidant) capacity is encountered to stop further proximal advance.
On a cellular level, neutrophils in the colonic epithelium can be thought of as microscopic H2O2factories, whose function can be replaced by exogenous H2O2. This interpretation is supported by rectal H2O2infusion studies in mice resulting in sharp inflammatory tissue delineation from normal tissue,contiguous inflammatory proximal extension, and rectal inflammatory persistence (discussed below),which are also characteristic of human UC[27]. Additionally, the colonic introduction of H2O2in humans results in classic UC[28]. Although this explains proximal extension, the next step is to explicate what causes these white blood cells (neutrophils) to move into the colonic epithelium in the first place causing inflammation and why it typically starts in the rectum?
Neutrophils are attracted into the colonic epithelium by H2O2 secreted by the colonic epithelium
Neutrophils are not the only cells in the body that produce H2O2. All living cells in the body generate H2O2from metabolic reactions, including colonic epithelial cells (colonocytes)[29]. Studies have shown increased production of H2O2in ascending non-inflamed colonic epithelium from patients with UC[30].This indicates a pre-inflammatory build-up of H2O2within colonocytes. In other words, H2O2builds up in colonic epithelial cells prior to the appearance of inflammation satisfying the absolute requirement of chronology for the cause (H2O2) to precede the effect (colitis).
H2O2is membrane permeable and can easily diffuse through the colonic epithelial cell membrane to the extracellular space[29]. Once outside the colonocyte, H2O2initiates inflammationviathe same mechanism as H2O2secreted by neutrophils,i.e.,oxidative disintegration of tight junctions and neutrophilic chemotaxis. Other studies have shown that reductive capacity (ability to neutralize H2O2)progressively decreases from proximal to distal regions of the colon with rectal epithelial cells having the least protection against the buildup of H2O2[31]. This causes the rectum to be the initial location in the colonic epithelium where H2O2will build up and, upon diffusion to the colonocyte extracellular space, attract neutrophils into the rectal epithelium causing inflammation and colitis. And due to its diminished reductive capacity, the rectum will be the last colonic region to heal resulting in a persistent ulcerative proctitis that is experienced by many patients.
Studies in genetically engineered mice that are unable to neutralize colonic H2O2[glutathione (GSH)peroxidase knock-out mice] develop colitis analogous to human UC[32]. This indicates that colonic epithelial cells can generate enough H2O2, which upon extracellular diffusion, can initiate colonic inflammation and colitis. The mechanism behind the initial increase in colonocyte H2O2giving rise to human UC will be discussed below in the section on oxidative stressors.
In other words, H2O2is a normal immune signaling molecule that attracts neutrophils. Neutrophils cannot determine which cell is secreting H2O2; whether it’s another neutrophil calling for help fighting an infection or a colonic epithelial cell leaking H2O2. In the latter case, neutrophils are simply doing what they are programmed to do given the stimulus (H2O2) they are exposed to. The development of UC indicates a healthy functioning innate immune system responding to a normal immune chemotactic factor (H2O2) being inappropriately secreted by the colonic epithelium. The correct treatment (discussed below) is not to abrogate this normal response with drugs that suppress essential innate normal immune reactivity but to restore colonic redox homeostasis so as to prevent colonocyte secretion of H2O2.
In summary, H2O2’s unique properties of cell membrane permeability, long life, potent oxidizing potential, and neutrophilic chemotactic capability combine to promote colonocyte extracellular diffusion followed by oxidative disintegration of colonic epithelial tight junctional proteins, which facilitates bacterial translocation from the colonic lumen into the sterile subjacent lamina propria while simultaneously (and chemotactically) attracting neutrophils into the colonic epithelium, both of which lead to colonic inflammation, and eventual UC (Figures 1A and 1B). H2O2initially accumulates in colonocytes and diffuses to the extra-cellular space in the rectal epithelium, which has the least tissue reductive capacity of the entire colon.
Neutrophils in the subjacent epithelial vasculature migrate along the interstitial H2O2concentration gradient to the source of the H2O2in the rectal epithelium. Once exposed to lumenal antigens,neutrophils are activated to secrete large amounts of H2O2, which promotes further neutrophilic infiltration while migrating the advancing edge of the inflammatory field to more proximal regions of the colon as described above. H2O2also causes vasodilation and severe damage to blood vessels with destruction of endothelial cells and disruption of endothelial cell tight junctions[33-35]. This leads to erythrocyte extravasation and bleeding into the colonic lumen as commonly observed in UC. Thus, the effects of H2O2on the innate immune system and vasculature explain both the microscopic and macroscopic features that characterize UC. The next section provides an evidence-based explanation for relapse. Following this, the concept of oxidative stress is discussed, which provides an evidence-based mechanism to explicate why H2O2builds up in the colonic epithelium to begin with.
Relapse: An acquired “hard-wired” vicious cycle of inflammation
Once complete histologic remission has been achieved and the colonic epithelium is free of inflammatory cells, neutrophils can, once again, migrate back into the colonic epithelium after medication is withdrawn. This resumption of inflammation after a period of quiescent disease is called relapse, also known as a flare. Stated differently, if UC were simply a function of exposure to environmental factors,neutrophils would not migrate back into the colonic epithelium causing mucosal inflammation and relapse after exposure has ceased and medication is withdrawn.
Relapse indicates that a fundamental change has occurred in colonic epithelial cells before and/or during mucosal inflammation leading to increased production of H2O2, which continues to diffuse throughout the extracellular space resulting in neutrophilic chemotactic migration into the colonic epithelium and eventual relapse. This is consistent with the significantly elevated intracellular colonocyte H2O2production observed in the non-involved colonic epithelium in patients with UC[30].The question is why do colonocytes in individuals with UC produce more H2O2than normal?
The answer is suggested by the susceptibility of mitochondrial DNA (mtDNA) to H2O2-induced oxidative damage. Due to their lack of histones, limited repair capability, and high single strand exposure time, mtDNA is highly susceptible to H2O2-induced oxidative damage[36,37]. H2O2induced oxidative damage to mtDNA introduces base mutations into the mitochondrial genome, which miscode during transcription of electron transport chain (ETC) complexes resulting in nucleotide mispairing and the incorporation of faulty protein subunits into the ETC. These acquired mitochondrial ETC mutations cause increased ETC electron leakage that produces increased amounts of superoxide, which is converted to excess H2O2. The end result is a dysfunctional mitochondrial ETC that generates higher levels of cellular H2O2, which upon extracellular diffusion initiates a relapse of colonic inflammation(Figure 1C)[38].
The elevated colonocyte H2O2resulting in mtDNA mutations originates from two sources. The initial increase in colonocyte H2O2is intracellular and originates from oxidative stress exposure (discussed in the next section). This is augmented by a large exogenous source of H2O2supplied by neutrophils that stream into the colonic epithelium and fill up the crypts of Lieberkühn. Being cell membrane permeable,H2O2can easily diffuse into surrounding epithelial stem cells and transition amplification cells, which give rise to the surface epithelium. This “back flow” of H2O2into colonocytes would ordinarily be neutralized by the cell. However, colonocyte reductive capacity has already been compromised by the initial rise in cellular H2O2due to oxidative stress exposure. This allows intracellular H2O2to diffuse unimpeded throughout the colonocyte into mitochondria leading to mtDNA oxidative damage and acquired mutations.
A causal role for mitochondrial ETC generated H2O2in the development of relapse is consistent with the onset of impaired mitochondrial beta-oxidation in the weeks leading up to relapse, which is reported to be caused by H2O2induced oxidative inhibition of mitochondrial thiolase, a necessary enzyme in the mitochondrial beta-oxidation pathway[39-41]. The involvement of ETC-generated H2O2in UC relapse is supported by reports of intractable UC in the setting of inherited ETC disfunction[42]. At birth, all mtDNA is normally identical. This is called homoplasmy. After H2O2-induced base mutations are introduced into the mitochondrial genome, all mtDNA is no longer identical. The simultaneous occurrence of genetically dissimilar cellular mtDNA (normal and mutated) is called mitochondrial heteroplasmy[43]. Studies have shown a significant degree of heteroplasmic mtDNA in the colonic epithelium of individuals with UC[44,45]. The presence of colonocyte mitochondrial heteroplasmy in UC will constitutively generate higher amounts of H2O2leading to additional mtDNA damage and greater H2O2production creating a self-amplifying vicious cycle of ever-increasing colonocyte H2O2[46].This constitutive internally reinforcing production of colonocyte H2O2perpetuates mucosal inflammation leading to relapse upon withdrawal of medication. The increased basal production of colonocyte H2O2promotes more frequent episodes of relapse and leads to refractory disease as colonocyte H2O2increases and UC becomes less responsive to medication.
Up until now, we have an H2O2-based mechanism that explains how UC begins, why inflammation extends proximally throughout the colon, and how mitochondrial heteroplasmy promotes a constitutive increase in colonocyte H2O2that contributes to relapse. What we are missing is why colonocyte H2O2becomes elevated in the first place. To understand this, we need to discuss the concept of oxidative stress as outlined in the next section for it is exposure to oxidative stress that initiatesde novodevelopment (and relapse) of UC.
OXlDATlVE STRESS
The invisible force that increases H2O2 and leads to UC
We are all subjected to oxidative stress since the moment of conception. But what is oxidative stress?More importantly, how can we define oxidative stress in a manner that is relevant for diagnosing disease, understanding pathogenesis, and advancing therapeutic intervention. Since most biological effects of reactive oxidant species are mediated by H2O2[47], and since cellular GSH is principally responsible for supplying the reducing equivalents (electrons) needed to neutralize H2O2[48,49], a clinical working definition of oxidative stress can be summarized as any stimulus that increases the amount or production of H2O2or elevates the risk of its occurrence by decreasing cellular reductive(antioxidant) capacity (i.e.,GSH). Stimuli that fulfill this definition are called oxidative stressors.

Figure 2 Redox homeostasis.Redox homeostasis is more than just a balance between oxidizing [hydrogen peroxide (H2O2)] and reducing agents (glutathione).In the above graph, redox homeostasis (slanted line) is maintained at both low and high H2O2 production rates (a and b), but the cell is functioning at a higher oxidative capacity (high capacity redox homeostasis) (b), when more H2O2 is being produced compared to times when lesser amounts of H2O2 are being generated(a). Mitochondria, the site of most cellular H2O2 production, do not synthesize their own glutathione and only contain 10% of the total cellular supply of this vital reducing equivalent that must be generated in the cytoplasm and imported into mitochondria, which takes time[53]. Once depleted, mitochondrial glutathione can take several hours to restore to normal levels[46]. In contrast to the limited supply of mitochondrial glutathione, studies have shown that mitochondrial electron transport chain production of H2O2 can increase up to 15 × during periods of high metabolic demand[54]. Any increase in H2O2 production forces the cell to utilize additional glutathione in order to maintain redox balance which may lead to high capacity redox homeostasis (b). Since about 30% of cell thiols (i.e., glutathione) normally undergo oxidation per hour[55], the additional oxidative stress imposed by high capacity redox homeostasis can, over time, deplete available glutathione and overwhelm colonocyte reductive capacity creating a state of impaired redox homeostasis (c) followed by H2O2 build-up and extracellular diffusion, which can lead to de novo ulcerative colitis or relapse. High capacity redox homeostasis is consistent with increased H2O2 production observed in the non-inflamed ascending colonic epithelium of individuals with ulcerative colitis[30]. H2O2: Hydrogen peroxide.
Oxidative stressors can be external (i.e.,environmental) or internal (originating in the body). Many oxidative stressors can be identified by the medical history and targeted for elimination by changes in diet and lifestyle. Clinically assessing the risk that oxidative stress will increase H2O2leading to worsening disease requires a working understanding of redox homeostasis. Redox homeostasis refers to the balance that is achieved when there is sufficient cellular reductive capacity (GSH) to neutralize the H2O2being produced. Thus, an oxidative stressor is a stimulus that places additional demands on the cell’s capacity to neutralize H2O2and maintain redox homeostasis. Over time, oxidative stress can disrupt the cell’s ability to maintain this critical balance. When this occurs it is called impaired redox homeostasis, which can lead to the build-up of colonocyte H2O2resulting in extracellular diffusion,mucosal inflammation, and UC as described above. Thus, identifying and eliminating oxidative stressors in order to assist in restoring colonic redox homeostasis is critical for the maintenance of longterm remission in UC. In order to maintain redox homeostasis, colonocytes utilize as much GSH as needed to neutralize the H2O2that is being produced by the cell. In this reaction, two molecules of GSH react with one molecule of H2O2viathe action of GSH peroxidase (GPx) to yield one molecule of GSH disulfide (GSSG) and one molecule of water, as illustrated below.
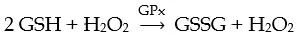
At higher levels of H2O2production significantly more GSH is consumed compared to lower levels of H2O2production in order to maintain a 2 (GSH) to 1 (H2O2) balance or redox homeostasis. However,higher levels of GSH consumption enhance the risk of GSH depletion after which free H2O2can begin to accumulate within the cell[50].
Thus, all redox homeostasis is not the same. Redox homeostasis maintained at high levels of H2O2production [high capacity redox homeostasis (HCRH)] increases the risk of GSH depletion and subsequent accumulation of intracellular H2O2(Figure 2). HCRH indicates that the cell has a greater capacity to oxidize substrate such as GSH and is consuming GSH at greater rates than normal. As long as the substrate being oxidized is GSH, redox homeostasis can be maintained and the cell is protected from the toxic effects of H2O2buildup. However, if GSH is depleted due to excessive utilization by HCRH, H2O2is free to diffuse throughout the cell and oxidize other molecules in the colonocyte such as enzymes and DNA. This can lead to metabolic disturbances such as impaired mitochondrial betaoxidation in addition to oxidative nuclear DNA mutations in tumor suppression and oncogenes that promote colorectal cancer as well as mtDNA mutations (mitochondrial heteroplasmy) that increase cellular H2O2production and facilitate disease relapse[30,51,52] (Figure 2).
Even if redox homeostasis is maintained during HCRH, the high demand for GSH may sequester this vital reducing agent away from other critical metabolic functions that depend upon GSH such as elimination of toxic xenobiotics and electrophiles, regulation of apoptosis and cell division, GSH dependent enzymes, maintenance of reduced vitamins C and E for cell membrane protection, and glutathionylation of proteins/enzymes to protect against irreversible oxidative damage[56-58]. Thus,HCRH may consume GSH needed for other cellular activity and compromise colonocyte and colonic functionality independent of the development of UC.
Exposure to multiple contemporaneous oxidative stressors facilitates progressively greater production of H2O2that increases the risk of reaching HCRH, which can lead to impaired redox homeostasis with the development of symptomatic UC (Figure 2C). This explains why the initial appearance of UC can be very explosive since HCRH may lead to sudden depletion of GSH causing significant acute increases in cellular H2O2, which can lead to severe mucosal inflammation as large amounts of colonocyte H2O2diffuse to the cell exterior. Lesser amounts of colonocyte extracellular H2O2can account for the pre-symptomatic systemic inflammation observed in individuals who go on to develop UC[59]. Seemingly insignificant oxidative stressors can lead to relapse for individuals functioning at HCRH. This should prompt a search for other unrecognized oxidative stress exposures to prevent relapse. Lastly, an important distinction to keep in mind is that oxidative stress is not the same as oxidative damage. Oxidative stress appears before oxidative damage occurs, while oxidative damage always indicates previous or ongoing oxidative stress. Rectal bleeding in UC is an indication of severe concurrent oxidative (H2O2induced) tissue damage caused by exposure to oxidative stress.
In summary, oxidative stress increases H2O2in the body. Oxidative stressors mediate the effects of oxidative stress on the body. As H2O2increases, colonocytes utilize greater amounts of GSH to maintain redox balance leading to HCRH, which can deplete cellular GSH leading tode novoUC or relapse. The increased H2O2production rates observed in the non-inflamed colonic epithelium of individuals with UC indicate the presence of HCRH[30]. The effects of multiple oxidative stressors are additive, each contributing to the cellular H2O2load. HCRH may lead to a GSH deficiency state and cellular disfunction.In the next section, we put it all together and derive the genetic susceptibility that predisposes to the development of UC.
PUTTlNG lT ALL TOGETHER
Predisposition, pathogenesis, pathophysiology, and relapse
Based on the cumulative data, we can now construct an evidence-based natural history of UC. The disease begins with exposure to one or more oxidative stressors, which increase the production of colonocyte H2O2. Over time, cellular reductive capacity is overwhelmed as the colonocyte is no longer able to maintain HCRH and H2O2accumulates in the cell. This is followed by H2O2diffusion through the cell membrane to the extracellular space within the crypts of Lieberkühn and the surrounding cellular microenvironment. This results in oxidative damage to interepithelial tight junctions and increased paracellular permeability accompanied by the creation of an H2O2diffusion gradient that envelops the subjacent microvasculature.
The combined effect of colonic epithelial antigenic translocation due to increased paracellular permeability and H2O2gradient-guided neutrophilic chemotaxis results in directed migration of neutrophils from the subjacent vasculature into the crypts of Lieberkühn along with the formation of neutrophilic cryptitis and crypt abscesses. Continued neutrophilic infiltration into the colonic epithelium leads to mucosal inflammation and UC (Figures 3A-E).
The rectum, having the lowest reductive capacity of the intestinal tract, is the initial site of H2O2accumulation and the first region to experience inflammation, which proceeds in a proximal direction as continuous secretion of large amounts of neutrophil released H2O2overcomes colonic epithelial reductive capacity in a circumferential manner resulting in a sharp demarcation between healthy and diseased tissue (epithelial cells at the same circumferential level have the same reductive capacity).Mucosal inflammation may reach more proximally and regress distally over time as a result of fluctuations in colonic epithelial reductive capacity due to changes in oxidative stress exposure (diet, stress,etc.) and epithelial repopulation.

Figure 3 Natural history of ulcerative colitis. The evidence-based natural history of ulcerative colitis begins with exposure to oxidative stressors (a), which increases colonocyte hydrogen peroxide (H2O2) (b). The increase in colonocyte H2O2 facilitates extracellular diffusion which overwhelms (oxidizes) local interstitial serum albumin antioxidant defense (60% of serum albumin is interstitial), leading to directed migration of neutrophils (chemotaxis) into the colonic epithelium (c) and mucosal inflammation (d) with subsequent development of ulcerative colitis (e). Large amounts of H2O2 are released by neutrophils into the extracellular space (f) with further oxidation of interstitial albumin and exhaustion of tissue antioxidant capacity (c). This worsens colonic inflammation (d) leading to local and systemic reductive depletion (i) as albumin is circulated through the colonic interstitium into tissue lymphatics and back into the systemic circulation. Neutrophil released H2O2 “back flows” into colonocytes (g) adding to the already elevated intracellular H2O2 levels resulting in mitochondrial DNA damage and mitochondrial heteroplasmy (b, red mtH). Mitochondrial heteroplasmy introduces mutations into the electron transport chain protein subunits, which generate additional H2O2 via enhanced electron leakage setting up a vicious cycle of ever increasing colonocyte H2O2 (b, red arrows). Increased colonocyte H2O2 diffuses into the extracellular space (h) causing disease relapse (c, d, e). The combination of local and systemic reductive depletion along with a ready supply of H2O2 from colonocytes and neutrophils (b and d)creates a mucosal inflammation that is self-amplifying, forward propagating, and auto-initiating (relapsing). Elimination of neutrophilic inflammation (d) by any means (i.e., immunosuppressive agents) will not stop relapse from occurring as colonocyte H2O2 continues to diffuse into the extracellular space (c, h). Conversely,normalizing colonocyte H2O2 alone will not stop the inflammation, which has become self-sustaining. This indicates that simultaneous elimination of all pathological sources contributing H2O2 to the inflammatory field must be achieved to ensure long-term remission and normal colonic functionality. Systemic reductive depletion may contribute to other serious health hazards as detailed below. H2O2: Hydrogen peroxide.
The large amount of H2O2released by neutrophils in the inflammatory field is chemotactic for other neutrophils in the subjacent vascular bed. This causes epithelial inflammation to become selfpropagating and auto-amplifying (Figure 3F), which enables prolonged contact between neutrophils and surrounding colonocytes. This close contact facilitates the back-flow of neutrophil-derived H2O2into adjacent colonocytes that adds to the already high colonocyte H2O2load due to oxidative stress exposure (Figure 3G).
High intracellular colonocyte H2O2promotes diffusion into mitochondria leading to mtDNA oxidation and the formation of acquired mtDNA mutations (mitochondrial heteroplasmy-mtH)(Figure 3B red arrows), which miscode during translation of ETC protein subunits. The resulting ETC mutations facilitate a higher degree of electron leakage and greater H2O2formation causing additional mtDNA damage and the creation of a vicious cycle, which maintains a high intracellular colonocyte H2O production that facilitates spontaneous relapse upon withdrawal of medication or exposure to everpresent oxidative stressors (Figure 3H).
Thus, based on the data, the derived genetic predisposition leading to UC is the inability of the colonic epithelium to cope with (neutralize) an oxidant (H2O2) load forcing the colonocyte into a state of impaired redox homeostasis after which free H2O2begins to accumulate in the cell (Figure 2C).Pathogenesis begins with impaired colonocyte redox homeostasis leading to the intracellular accumulation of H2O2after HCRH has exhausted cellular reductive capacity (Figure 2C). The pathophysiology commences with the influx of neutrophils into the colonic mucosa, which defines the beginning of what will eventually become the symptomatic phase of the illness. However, extracellular colonocyte diffusion of H2O2does not inexorably lead to UC due to the presence of a secondary “back-up” system of antioxidant defense provided by human serum albumin (HSA) (Figure 3C) as discussed in the following section.
HSA: The link between colonocyte and systemic redox homeostasis
Although UC is traditionally thought of being limited to the colon, the significant decrease reported in total blood antioxidant capacity (erythrocytes and plasma) in individuals with UC suggests that excess colonic production of H2O2is causing impaired systemic redox homeostasis as well[60-62]. In other words, the capacity of the blood to remove H2O2in UC patients is compromised. Why would colonic production of H2O2affect systemic redox homeostasis and what effect might this have on the severity of UC and overall patient health?
Systemic redox homeostasis is provided by HSA and erythrocytes, both of which are highly effective scavengers of H2O2. Their combined action maintains blood H2O2at very low levels, in the range of 0.8-6µM for healthy individuals[63]. HSA can directly scavenge H2O2viaa reduced surface cysteine thiol(cys34)[64]. In addition, the HSA molecule itself has a GSH-linked thiol peroxidase activity that can remove circulating H2O2[65]. The significant anti-oxidant scavenging ability of HSA represents approximately 70% of the free radical trapping ability of human serum[64]. Since 60% of HSA is present in the interstitial space, this indicates that HSA acts as an extracellular backup anti-oxidant defensive layer(after intracellular colonocyte GSH) that protects against the development of UC by preventing the interstitial accumulation and diffusion of colonocyte released H2O2from reaching the subjacent epithelial blood vessels and attracting neutrophils into the colonic epithelium leading to colonic mucosal inflammation and UC. Interstitial albumin has a turnover of 4% per hour after which it recycles back to the systemic circulation[66]. This suggests that each day the entire blood supply of reduced albumin is exposed to excess colonic H2O2with a significant portion becoming oxidized.
Red blood cells also provide a significant level of systemic anti-oxidant defense. Erythrocytes contribute up to 30% of whole blood reductive capacity. Erythrocytes are highly permeable to H2O2[67].With a normal mean inter-erythrocytic distance of 3 µm, a molecule of H2O2in the circulation will encounter a red blood cell before it encounters HSA[68]. Free serum H2O2will diffuse into red blood cells where it is neutralized by significant anti-oxidant enzymatic defenses comprised of catalase and GSH[67]. Whereas H2O2released by colonocytes or infiltrating epithelial neutrophils during active UC can oxidize HSA in the interstitial space, decreased red blood cell reductive capacity (i.e.,decreased erythrocyte GSH) implies that colonic H2O2is diffusing directly into the systemic circulation and into erythrocytes with depletion of total blood reductive capacity. The inability of interstitial HSA to completely remove colonic H2O2will lead to H2O2accumulation and a greater degree of tissue damage accompanied by neutrophilic infiltration resulting in increased severity of disease, which contributes to relapse (Figure 3F). This is supported by studies showing that the loss of blood reductive capacity(inability to remove H2O2) is associated with worsening UC[62]. The critical role of reduced albumin for the maintenance of colonic interstitial redox homeostasis is illustrated by studies showing that the onset and progression of experimental murine colitis were prevented by reduced (reductively enhanced)albumin, which, in turn, was strongly associated with an improved systemic reductive capacity[69].
Reduced albumin also directly neutralized H2O2and prevented thein vitroloss of tight junctional proteins in human intestinal cell tissue treated with H2O2[69]. This indicates that reduced albumin can act as an interstitial reducing agent (antioxidant) and delay/prevent the onset of UC by neutralizing interstitial H2O2released by colonocytes before the H2O2can initiate chemotactic directed migration of neutrophils into the colonic epithelium. This implies that oxidized, or decreased albumin levels would offer less protection and hasten the development of UC. This is consistent with worsening UC observed in association with anemia and hypoalbuminemia[70,71]. Other studies report a significant inverse relationship between low serum albumin and risk of colectomy[72]. Conversely, studies in UC patients have shown that mucosal healing is positively associated with high (> 4.4 mg/dL) serum albumin[73].
Although low HSA can be secondary to colonic protein loss, and improved HSA levels may follow mucosal healing, the association of high HSA levels with a protective effect in conditions without colitis such as Bell’s palsy and coronary artery disease implies an independent association with an intrinsic property of HSA such as oxidation status and not just as a biomarker for worsening colitis[74,75]. Thus,it is reasonable to assume that it is not just worsening colitis that causes low albumin but low serum albumin reductive capacity causing worsening colitis that increases albumin loss. HSA oxidation status should be part of screening lab work as oxidized albumin is inherently proinflammatory and associated with the progression of other diseases in addition to UC[76,77]. The proinflammatory nature of oxidized albumin and subsequent loss of systemic reductive capacity may be reflected in the worsening health and the high healthcare resource utilization in the year leading up to the diagnosis of UC[78]. This insight provides a critical therapeutic window of opportunity to restore systemic redox homeostasis and prevent UC if HSA is found to be significantly oxidized. In summary, once symptomatic UC develops,local and systemic reductive depletion is likely. At this point, colonocytes have already been exposed to genotoxic levels of H2O2for many months or years setting the stage for molecular oxidative alterations that lead to life-long relapse (Figures 3A-H). However, the oxidative nature of these changes offers the possibility of reversal with a therapeutic reducing agent (detailed in treatment section below).
HOW OXlDATlVE STRESSORS GENERATE H2O2 lN THE BODY
Oxidative stress: Sources, classification, and definition
H2O2is produced by many different cellular enzymatic reactions. Using the advanced search option in the BRENDA enzyme database limited to “homo sapiens” as the organism and “H2O2” as the product in the subitem text field returned 29 different enzymes acting upon 188 distinct molecular substrates[79].This does not include non-enzymatic reactions such as the auto-oxidation of oxyhemoglobin or subunits of the mitochondrial ETC, which is considered the principal source of H2O2in the body. Oxidative stressors are extremely diverse in their mechanism of action with some increasing the substrate for a single enzyme while others can affect every H2O2generating system in the body by inhibiting critical anti-oxidant enzyme systems needed for H2O2removal. Individuals with UC are usually contemporaneously affected by more than one oxidative stressor but the commonality among all oxidative stressors,however, is the production of H2O2. Consequently, oxidative stressors are additive since they all increase the H2O2load in the body. This may cause different individuals with UC to be more or less affected by the same oxidative stressor depending on the pre-existing H2O2load in the colonocyte and state of redox homeostasis, which can change over time (Figure 2).
This can also result in the individual tolerance for the same oxidative stressor to vary with age,comorbidity, lifestyle and exposure duration and intensity. Thus, based on the evidence, it is reasonable to conclude that all factors that increase the risk of relapse or developingde novoUC are oxidative stressors that increase H2O2in the body. Conversely, all oxidative stressors are risk factors for relapse or the development of UC. A convenient method of classification is grouping oxidative stressors as exogenous (originating external to the body) or endogenous (originating inside the body). The following section details the mechanism of action for several reported and/or common oxidative stressors associated with UC.
Exogenous oxidative stressors
Diet has been implicated in the pathogenesis and pathophysiology of UC[80]. 65% of surveyed individuals with UC believe that food is a significant trigger for relapse with 50% asserting that diet contributed to the initial development of disease[81]. This suggests that dietary factors exert their effect on the pathogenesis of UC by means of a common mechanism within the molecular chain of events leading to the onset of disease.
Dietary fat
Studies have shown that a high-fat low-fiber “westernized” type diet is associated with the development of UC, and high-fiber low-fat diets reduce systemic inflammatory biomarkers in patients with this illness[82,83]. Additionally, diets high in total fat and certain fatty acids are associated with exacerbation of UC[82,84,85]. But how does dietary fat initiate or worsen UC? Peroxisomes play an indispensable role in the metabolism of fatty acids obtained from dietary fat[86]. Peroxisomes are involved in the metabolism of dietary lipids such as medium chain, long chain, and very long chain fatty acids and cholesterol in addition to pristanic and phytanic acids[86,87]. Peroxisomal metabolism of fatty acids generates large amounts of H2O2, which is estimated to be about 35% of total cellular H2O2production[88]. This is in line with data implicating peroxisomal H2O2as an important source of cellular oxidative stress[89]. This implies that excess peroxisomal generated H2O2can overwhelm the cell’s reductive (antioxidant) capacity and accumulate to the point of causing cellular dysfunction. This is consistent with previous data ascribing a causal role for H2O2in the pathogenesis of UC and implies that high fat diets contribute to the pathogenesis and relapse of UC by generating large amounts of peroxisomal H2O2. Excess peroxisomal generated H2O2can diffuse into the cytoplasm and overwhelm the colonocyte’s ability for its removal leading to extracellular diffusion and the development or relapse of UC as described above. Thus, high fat diets are risk factors for the development of UC because they are oxidative stressors that generate large amounts of peroxisomal H2O2[89]. Low fat diets ameliorate colonocyte oxidative stress by decreasing production of peroxisomal H2O2and in so doing promote remission of UC.
Fiber
As mentioned above, low fiber diets are associated with the development and worsening of UC. But how does fiber interface with the pathogenesis and relapse of this illness? The colonic epithelium utilizes short chain fatty acids (i.e.,butyrate) for most of its energy requirements[90]. The production of butyrate starts with the fermentation of dietary soluble fiber by colonic bacteria. Butyrate is rapidly absorbed by colonic epithelial cellsviapassive diffusion and cell membrane transport proteins[91]. Once in the cytoplasm, butyrate is transportedviathe carnitine shuttle into mitochondria where it undergoes beta-oxidation. The resulting acetyl-coenzyme A (CoA) enters the Krebs cycle, which generates reducing equivalents (NADH, FADH2) that provide the energy for oxidative phosphorylation and ATP production[92]. This process provides up to 70% of colonocyte energy supplies (Figure 4A)[93,94].
A decrease in the available dietary soluble fiber will diminish the amount of butyrate absorbed by colonic epithelial cells and less butyrate will be available to undergo mitochondrial beta-oxidation. With decreased beta-oxidation of butyrate generating less acetyl-CoA, the colonocyte may not have enough fuel for the Krebs cycle to produce sufficient reducing equivalents (NADH, FADH2) in order to power oxidative phosphorylation and provide the energy for the biosynthesis of ATP. Without sufficient ATP to fuel critical cellular functions, the colonocyte will die. To increase ATP production, the colonocyte diverts glutamate into the Krebs cycle (viaalpha keto-glutarate) in order to replace Krebs cycle intermediary metabolites that would otherwise be supplied by dietary fiber, which is in low supply(Figure 4B). Glutamate (an amino acid) is derived from the amino acid glutamine (the storage form of glutamate), and studies have shown cellular diversion of glutamine into the Krebs cycle as a consequence of impaired mitochondrial pyruvate transport underscoring glutamine’s role as a backup energy supply during times of limited acetyl-CoA availability[95].

Figure 4 Normal vs low-fiber colonocyte bioenergetics. A: The normal vectorial bioenergetic flux beginning with soluble dietary fiber that is converted to short-chain fatty acids (i.e., butyrate) by bacteria in the colonic lumen. Butyrate is rapidly absorbed by colonic epithelial cells (colonocytes). Once inside the colonocyte, butyrate undergoes mitochondrial beta-oxidation to generate acetyl-coenzyme A (CoA), which is processed by the Krebs cycle that produces NADH and FADH2. The high-energy electrons present in NADH and FADH2 are used to drive oxidative phosphorylation (Oxphos) resulting in the biosynthesis of ATP, which fuels most of the cell’s energy needs; B: Low fiber intake decreases available butyrate needed for acetyl-CoA production. Under these energy-restricted conditions,glutamate is diverted into the Krebs cycle and away from the synthesis of glutathione (GSH). Diversion of glutamate into the Krebs cycle is called anapleurotic metabolism (red curved arrow) and is needed to replenish depleted Krebs cycle intermediary metabolites that would otherwise be supplied by dietary soluble fiber,which can no longer perform this role due to a low fiber diet. Since glutamate is needed for the synthesis of GSH, the sequestration of glutamate as a replacement energy source restricts the amount of glutathione the cell is able to synthesize. GSH is the principal reducing equivalent required to neutralize cellular hydrogen peroxide (H2O2). Insufficient glutathione will cause cellular H2O2 to increase, which upon extracellular diffusion may initiate neutrophil chemotaxis into the colonic epithelium and de novo ulcerative colitis or disease relapse. Interruption of colonocyte bioenergetic flux anywhere along the pathway from the microbiome to acetyl CoA will increase colonocyte anapleurotic metabolism and cellular H2O2, which can lead to ulcerative colitis. H2O2: Hydrogen peroxide; CoA: Coenzyme A.
Studies on isolated colonocytes from germ-free rats (that cannot produce butyrate) report a 45%increase in glutamine use by these cells compared to conventionally reared animals[96]. Other studies using isolated colonocytes from germ-free mice demonstrated 16-fold lower NADH/NAD+ ratios as well as 56% lower ATP levels[97]. Colonization of germ-free mice with flora from conventional mice or butyrate-producing bacteria rescued the colonocyte energy deficit as did butyrate exposure to isolated colonocytes from germ-free mice[97]. This indicates that butyrate is a critical source of energy for colonocyte ATP production and colonocytes will compensate for the loss of butyrate by diverting glutamine (viaglutamate) into the Krebs cycle to maintain the production of ATP.
However, the continued Krebs-cycle oxidation of glutamine as a backup energy source (i.e.,during prolonged low-fiber diets) entails significant consequences for the colonocyte. Glutamine is the precursor to glutamate, which is necessary for GSH synthesis. The diversion of glutamate into the Krebs cycle (called anapleurotic metabolism) to sustain cellular energy requirements restricts glutamate’s availability for the biosynthesis of GSH, which is critical for the elimination of cellular H2O2[98]. Studies have shown that disrupting mitochondrial pyruvate uptake directs glutamine into the Krebs cycle and away from GSH synthesis[99]. Because GSH is critical for the elimination of cellular H2O2, a decrease in GSH synthesis will lead to increased colonocyte H2O2and subsequent diffusion through the cell membrane to the extracellular space, which may precipitatede novoUC, worsen existing UC, or contribute to relapse (Figure 1B). Thus, low fiber diets are oxidative stressors because they increase the risk of colonocyte intracellular H2O2buildup.
The critical importance of (soluble) dietary fiber for colonic bioenergetics and redox homeostasis is underscored by what occurs with the complete absence of colonic fiber. Diversion colitis is a reactive colonic inflammatory response in the by-passed segment of the large intestine as a result of fecal stream diversion secondary to colostomy or ileostomy. Under these circumstances, there is no dietary fiber entering the defunctioned segment of the large intestine. This results in a colitis affecting nearly all individuals undergoing this procedure within 1 to 3 years after colonic diversion[90]. Histopathology soon after onset shows an influx of neutrophils into the colonic epithelium (analogous to UC)[100]. This suggests the alternate use of glutamate to compensate for the complete lack of lumenal butyrate decreases colonocyte GSH enough to raise cellular H2O2to levels that facilitate extracellular diffusion and the development of colitis. It also implies that H2O2plays a prominent causal role in the development of diversion colitis as well[101].
This interpretation is consistent with a case report of a healthy 36-year-old male who developed UC after following an extremely low carbohydrate diet for weight loss, which resolved without medication upon the institution of a semi-vegetarian diet[102]. Studies have demonstrated significant declines in fecal butyrate and butyrate-producing bacteria in individuals with reduced dietary carbohydrates[103].The decrease in colonocyte butyrate can be reversed with butyrate enemas that significantly increased colonic epithelial GSH, which is consistent with a GSH sparing effect of butyrate inferred from Figure 4B[104].
Butyrate enemas also significantly reduced mucosal inflammation in patients with refractory UC[105]. This can be attributed to the butyrate-mediated increase in colonocyte GSH and subsequent reduction in colonic epithelial H2O2. This is supported by studies showing that butyrate prevents H2O2-induced DNA damage in isolated human colonocytes[106]. Thus, colonic butyrate has a critical role in maintaining colonocyte redox-homeostasis by preventing the anapleurotic metabolism of glutamate and subsequent decrease in colonic epithelial GSH, which leads to elevated colonocyte H2O2and UC[107].
The crucial role of GSH in the elimination of cellular H2O2can be seen in GPx knockout mice that lack this key enzyme needed to utilize GSH for the elimination of H2O2. Knockout mice lacking GPx develop colitis[32]. Colitis also occurs concomitantly with experimental beta-oxidation inhibition in mice, and in pigs subsequent to vitamin B-5 (pantothenic acid) deficiency. Vitamin B-5 is necessary for CoA synthesis, without which there is no acetyl CoA[108,109]. This suggests that disruption of bioenergetic flux at any point along the metabolic pathway from lumenal fiber to the formation of acetyl CoA will result in increased colonocyte H2O2and colitis (Figure 4B).
Based on these data, we can reasonably predict that alterations in the colonic flora (microbiome dysbiosis) that lead to impaired short-chain fatty acid (i.e.,butyrate) production will contribute to the development and relapse of UC by increasing colonocyte H2O2. A diverse set of adverse environmental exposures can shift the colonic microbiome towards dysbiosis and impaired butyrate production. These include high fat/low fiber/high protein diets, food additives in processed food, smoking and alcohol ingestion[110-113]. Other diverse factors such as infant delivery and feeding methods, medications,enteric endocrine disruptors, psychological stress can also facilitate microbiome dysbiosis[114-117].Long term dietary patterns that include soft drinks and artificial sweeteners may tip the balance towards dysbiosis[118,119]. Thus, microbiome dysbiosis is an oxidative stressor that can increase colonocyte H2O2and contribute to the onset of UC[120].
Lastly, as mentioned above, H2O2, being membrane permeable, can diffuse into the colonocyte nucleus leading to oxidative nuclear DNA mutations in tumor suppression and oncogenes that promote colorectal cancer[51,52]. Studies have shown that mice fed a total western diet develop a neutrophil predominant colitis and colorectal cancer[121]. This is analogous to histological findings in human UC,which also carries an enhanced risk of colorectal cancer. Taken together, when the evidence supporting a causal role for H2O2in UC and its associated colon cancer is viewed in light of biological mechanisms leading to increased colonocyte H2O2subsequent to low-fiber high-fat diets, it is reasonable to conclude that the increased incidence of UC and colorectal cancer associated with the (low-fiber high-fat) western diet[9,97,122] is mediated through elevated colonocyte H2O2. This raises the possibility of primary preventionviachanges to reduce dietary oxidative stress (i.e.,high fiber, low fatetc) and/or the administration of an oral reducing agent (detailed in the treatment section below).
Alcohol
Several studies have found that alcohol consumption increases the risk of onset, relapse, and gastrointestinal symptoms in individuals with UC[123]. Alcohol is biomembrane permeable, and after ingestion is distributed to all tissues in the body[124]. Alcohol metabolizing enzymes in the colonic epithelium can generate large amounts of H2O2[124]. Alcohol metabolism by alcohol dehydrogenase generates acetaldehyde, which is converted to acetic acid by aldehyde dehydrogenase. Both these reactions generate NADH, which feeds into the mitochondrial ETC causing increased electron leakage and enhanced generation of H2O2[124]. The increased amount of colonocyte H2O2can overwhelm cellular reductive capacity and diffuse to the extracellular space leading to relapse or the onset of UC.
Cytochrome P4502E1 (CYP2E1) is a second alcohol oxidizing enzyme that is highly expressed in the human intestine and is upregulated by chronic alcohol exposure[125]. CYP2E1 has the highest catalytic activity among the members of CYP enzymes in metabolizing ethanol[126]. CYP2E1 consumes NADPH when metabolizing ethanol to acetaldehyde and in the process generates large amounts of H2O2[126,127]. NADPH is also required for the recycling of oxidized GSH (GSSG) back to reduced GSH by GSSG reductase[49]. This can reduce the availability of GSH for the elimination of H2O2and contribute to increased colonocyte H2O2levels. The total combined effect of alcohol metabolism is excess production of colonocyte H2O2that can overwhelm cellular reductive capacity leading to extracellular H2O2diffusion andde novoUC or relapse by the mechanisms detailed above. CYP2E1 is also upregulated by ethanol, which magnifies the oxidative stress caused by this alcohol metabolizing enzyme.
Antibiotics
Due to their widespread use, antibiotics represent a significant source of oxidative stress within the population. Studies have shown an association between antibiotic use and the development of UC[128].Although antibiotics are administered to eradicate pathogenic bacteria they also indiscriminately kill beneficial commensal bacteria that make up the colonic microbiome leading to a decrease in species diversity including a reduction in bacterial species that produce butyrate[129]. A decrease in colonic butyrate can lead to metabolic changes favoring increased colonocyte production of H2O2(Figure 4).Antibiotic-induced microbiome depletion (dysbiosis) can last for years and act cooperatively with other oxidative stressors such as a high-fat diet[130,131]. This can hasten colonocyte H2O2build-up (HCRH)and the development of UC.
However, microbiome depletion is not the only mechanism by which antibiotics generate excess colonocyte H2O2. Studies have shown that antibiotics induce the production of significant amounts of H2O2in both bacteria and human intestinal epithelial cells, which in the latter was caused by an alteration to the ETC[132-135]. Since H2O2is cell membrane permeable, bacterial H2O2can diffuse across epithelial cell membranes and add to the already increased antibiotic-induced colonic epithelial cell H2O2load.This can lead to mtDNA oxidative damage with the formation of colonocyte mitochondrial heteroplasmy and ever-increasing production of intracellular H2O2resulting in HCRH (Figure 2). Increased cellular production of H2O2can overwhelm colonocyte reductive (antioxidant) capacity and lead to a buildup of colonocyte H2O2that will facilitate the development of UC years later. Because virtually everyone is exposed to antibiotics at one time or another, they exert a selective oxidative pressure that can manifest as UC in individuals with a predisposing genetic makeup encoding for a diminished reductive capacity that facilitates the buildup of H2O2.
Psychological stress: A common oxidative stressor leading to H2O2 production
Stress is a significant risk factor for UC. Up to 40% of patients with UC report psychological stress as an exacerbating factor[136]. Psychological stress can causede novoUC and worsen existing disease[137-141]. Psychological stress exposure is reported to induce mucosal inflammatory responses and can result in colonic hypermotility that may be sufficient to occlude the lumen[142,143]. But why is psychological stress pro-inflammatory in the colon and how does stress initiate or worsen UC?
The coordinated movement of food along the gastrointestinal (GI) tract is dependent on 5-hydroxytryptamine (serotonin) mediated regulation of smooth muscle tone, motility, and peristalsis[144]. 95% of serotonin is stored in enterochromaffin cells (EC) that are present in the GI tract mucosa[145]. Serotonin is released from EC cells and stimulates enteric nerve terminals to initiate a peristaltic wave[144,146].However, the amount released is much more than needed and the excess serotonin is rapidly taken up by colonic epithelial cells and metabolized by colonocyte monoamine oxidase (EC#1.4.3.4)[144]. This prevents hyper-stimulation and excessive bowel motility that can lead to colonic spasms. Mono-amine oxidase catalyzes the oxidative deamination of serotonin in a process that generates H2O2; the reaction catalyzed is RCH2NHR’ + H2O + O2→ RCHO + R’NH2+ H2O2[147].
Studies have shown that psychological stress causes prolonged increases in colonic motility[148].Stress-induced colonic hypermobility and spasm will release large amounts of serotonin into the colonic mucosa that is metabolized to H2O2within colonocytes. The excess colonocyte H2O2can acutely overwhelm the enterocyte’s antioxidant capacity resulting in H2O2accumulation and eventual UC after extracellular diffusion. This mechanism is supported by studies showing that serotonin has a key role in the pathogenesis of experimental colitis[149].
Thus, psychological stress has a pernicious effect on the course of UC but UC is also psychologically stressful with studies reporting that patients with UC are engaged in a continuous “fight” to maintain health-related normality[150]. This sets up a self-sustaining bidirectional cycle of continuous psychological stress that contributes to increased frequency and severity of disease[151]. Although stress can cause or worsen UC, stress reduction is generally not effective at altering the activity or course of disease sufficiently to induce remission[152,153]. This is not surprising since the principal driving force behind the auto-propagating nature of inflammation in UC is H2O2release by activated infiltrating mucosal neutrophils and not the metabolism of serotonin, which has its principal effect as a contributing factor in the stress-induced pathogenesis and relapse of UC[26,149].
Cigarette smoking: Releasing the brakes
Since reports in the early 1980s, numerous studies included in three meta-analyses (1989, 2006 and 2021)have established that cigarette smoking is significantly protective against the development of UC compared to non-smokers while smoking cessation is a significant risk factor for developing UC or experiencing disease relapse with increased severity of illness[154-158]. Additionally, as noted below,cigarette smoking also affects the therapeutic response after smoking cessation. But why is cigarette smoking protective against the onset of UC?
Nicotine, an addictive chemical present in tobacco, was initially considered as a possible protective factor. Nicotine is effective at inducing remission when begun at the time of or soon (up to 4 wk) after smoking cessation (early relapse)[159,160] However, nicotine is largely ineffective when therapy is administered for disease relapse several months or years after smoking cessation (late relapse) with studies concluding that nicotine therapy is of minimal value in the treatment of UC and questions whether nicotine is the active protective component of smoking that decreases risk and inflammation in UC[161-164]. In contrast, the resumption of smoking is reported to be an effective therapy for induction and long-term maintenance of remission in patients with early or late relapse[159,160,165]. Indeed,studies have reported that resumption of smoking is highly effective for induction of remission in refractory disease years after smoking cessation; with nicotine being effective if treatment is begun at the same time as smoking cessation[160]. Thus, in ex-smokers, nicotine is effective for remission induction in early relapse while the resumption of smoking is effective in both early and late relapse.
Taken together, the data indicate that there are two distinct oxidative stressors with distinct short and long-term mechanisms of action both of which are caused by the latent and repressed effects of active smoking and are only unmasked by smoking cessation. The initial mechanism manifests soon after smoking cessation and lasts for days to under a month. In contrast, the second mechanism becomes predominant many months to years later reaching a peak of the highest risk of relapse within 2 to 5 years of smoking cessation[166].
The initial oxidative stressor is the physiological and psychological stress of nicotine withdrawal,which is manifest shortly after smoking cessation and peaks within the first week, and lasts up to one month[167]. Nicotine withdrawal symptoms can include anger, irritability, frustration, anxiety,depression, insomnia, restlessness, and constipation[167]. These same psychological emotions of anger,resentment, emotional conflict, hostility, anxiety, and psychological tension were observed under direct observation to cause significant colonic hypermotility and spasm[168]. Thus, it is reasonable to conclude that nicotine withdrawal secondary to smoking cessation can result in colonic hypermotility with increased colonic serotonin secretion and enhanced monoamine oxidase production of H2O2(see above section-psychological stress). Under these circumstances, the administration of nicotine will decrease colonic hypermotility and lower colonic serotonin production, which decreases colonocyte H2O2leading to remission. Resumption of cigarette smoking also provides the nicotine needed to treat early relapse due to nicotine withdrawal.
In other words, nicotine-induced remission is due to the relief of nicotine withdrawal symptoms (and accompanying colonic hypermotility) during early relapse after smoking cessation. Nicotine treatment is rendered minimally effective after nicotine withdrawal symptoms (and colonic hypermotility) have subsided. Colonic hypermotility (from any cause) is an oxidative stressor that increases colonocyte H2O2, which can overwhelm colonocyte reductive (antioxidant) capacity leading to extracellular diffusion and UC.
The second oxidative stressor caused by cigarette smoking cessation is due to disinhibition of the colonocyte ETC. Studies quantifying the effect of cigarette tar on mitochondrial ETC activity report an 82% inhibition rate on whole chain respiration[169]. Under normal conditions, the ETC is fueled by electron flux provided by reducing equivalents (NADH and FADH2) generated by the multi-enzyme Krebs cycle[170]. H2O2is produced by spontaneous auto-oxidation of the ETC (electron leakage). These leaked electrons combine with vicinal oxygen within the mitochondrial matrix to form superoxide,which is converted to H2O2by superoxide dismutase. H2O2is subsequently neutralized by GPx using GSH as a reducing co-factor.
Under conditions of ETC inhibition during active smoking, less ETC-generated H2O2is produced,which affords protection against the development of UC. However, while smoking, upregulation of bioenergetic enzyme systems can be expected as the colonocyte attempts to overcome the smokinginduced inhibition and restore normal mitochondrial bioenergetics[171]. Upon smoking cessation, the inhibition is slowly lifted and increased production of ETC “fuel” is metabolized producing supraphysiological amounts of H2O2as a result of increased ETC auto-oxidation (electron leakage).Colonocytes respond to this oxidative stress by producing additional GSH for H2O2neutralization. This creates a condition of HCRH (Figure 2) that can eventually overwhelm colonocyte reductive capacity leading to cellular H2O2build-up and eventualde novoUC as H2O2diffuses to the extracellular interstitial space attracting neutrophils into the colonic epithelium from the subjacent microvasculature.
Smoking resumption inhibits ETC activity, which decreases colonic epithelial cell H2O2leading to remission. Nicotine does not inhibit the ETC and cannot induce remission of late UC relapse (years after smoking cessation). This interpretation is supported by studies that demonstrated significantly improved clinical manifestations such as bloody stool, diarrhea, and abdominal pain in UC patients treated with metformin[172]. Further, UC patients treated with metformin showed a significant decrease in histological and endoscopic disease scores in addition to significantly diminished erythrocyte sedimentation rate (a biomarker of systemic inflammation) and significantly decreased indices of colonic local oxidative stress (tissue malonaldehyde and myeloperoxidase)[172].
But how does metformin improve UC and how is it related to cigarette smoking? Metformin is a biguanide antihyperglycemic agent used to treat type 2 diabetes. Its mechanism of action includes inhibition of mitochondrial glycerol 3-phosphate dehydrogenase (of the glycerolphosphate shuttle-EC 1.1.5.3) and inhibition of ETC complex I both of which are major contributors of electrons to the ETC in mitochondria[173-175]. Inhibition of electron flux by metformin is analogous to the ETC inhibitory effects of cigarette smoking. This implies that both smoking and metformin improve UC by decreasing mitochondrial production of H2O2. This is supported by studies showing that targeted inhibition of glycerol 3-phosphate dehydrogenase decreases cellular production of H2O2[176].In summary, the evidence supports two distinct oxidative stress mechanisms to explain the effects of smoking cessation on UC. Early relapse after smoking cessation (within days) is mediated by the oxidative stress induced by the physiological effects of nicotine withdrawal while late relapse (months to years) is mediated by a slow rise in colonocyte H2O2due to disinhibition of mitochondrial H2O2-generating metabolic pathways. Conversely, cigarette smoking affords protection against early and late UC relapse by providing nicotine and decreasing colonocyte production of H2O2, respectively.
Mercury
Mercury is a major environmental contaminant and a significant source of occupational exposure[177].Occupational inhalation of mercury vapor is reported to cause a recurrent relapse of UC[178]. Mercury forms stable bonds with thiol groups present on GSH in addition to inhibiting GPx, both of which are critical for the removal of cellular H2O2[179-181]. This results in the inactivation of the entire GSH-based antioxidant system. The compromise of this critical system by mercury prevents neutralization of H2O2,which can accumulate to excessive levels inside colonocytes leading to extracellular diffusion and UC relapse as described above. Thus, mercury is an oxidative stressor that increases cellular H2O2by preventing its elimination from the cell. Mercury is a pervasive contaminating xenobiotic whose exposure is likely to be insidious, bio-accumulative, and additive to other contemporaneous oxidative stress exposures.
PERFLUOROOCTANOlC AClD
Perfluorooctanoic acid (PFOA) is a ubiquitous environmental contaminant that was used to manufacture non-stick pans in addition to other commercial products such as stain and water-resistant fabrics. Introduced into the environment in the 1950s, PFOA can be found in the serum of virtually all residents of industrialized countries. Human exposure occursviamany sources including contaminated drinking water, food, and house dust. Due to the high dissociation energy of its carbon-fluorine bond,PFOA is resistant to vertebrate metabolism and environmental degradation[182]. As a result, PFOA is called a “forever-chemical” because it is not biodegradable and has a long elimination half-life of 3.5 years[183].
Studies have demonstrated a significant exposure-response relationship between PFOA serum levels and subsequent UC but no association with Crohn’s disease (CD)[183]. Other studies have reported significantly increased serum PFOA in UC patients compared to a combined group of CD (positive control for intestinal inflammation) and normal individuals (negative non-diseased control)[184]. The specific association of PFOA with UC suggests that PFOA’s effect is acting in concert with a unique predisposing genetic makeup to select a subset of individuals for the development of UC. In other words, PFOA’s mechanism of action in all exposed individuals is the same but the genetic predisposition in a subset of individuals is permissive for the development of UC.
A related halogenated chemical, 2-bromooctanoic acid, after conversion to the sodium salt 2-bromooctanoate, is reported to cause an acute murine colitis analogous to human UC after rectal installation[108]. This suggests the possibility that PFOA might be acting in the same manner as 2-bromooctanoate since halogenated carbon compounds, as a group, have a high resistance to degradation[185].
In human UC, beta-oxidation is inhibited as a secondary effect caused by rising levels of colonocyte H2O2[30]. Since 2-bromooctanoate causes murine UC accompanied by inhibition of beta-oxidation and the related halogenated chemical PFOA causes UC and is not biodegradable this suggests that intracellular H2O2is increased as a result of the colonocyte’s high expenditure of energy (ATP) in a futile attempt to metabolize and remove these non-biodegradable halogenated xenobiotics from the cell. Since almost all cellular ATP is produced as a result of mitochondrial ETC activity, which also generates most of the cell’s H2O2, this implies that the initial buildup of H2O2occurs in mitochondria where betaoxidation is also located. Increased mitochondrial H2O2will inhibit mitochondrial thiolase, the last enzyme in the beta-oxidation cascade, leading to inhibition of mitochondrial beta-oxidation[30].
In other words, the colonocyte’s persistent metabolic response in an attempt to eliminate these nonbiodegradable chemicals leads to increased H2O2generated by the ETC, which inhibits mitochondrial beta-oxidation followed, sometime later, by UC as H2O2diffuses out of the colonocyte into the extra cellular space. This mechanism is consistent with studies showing inhibition of beta-oxidation in UC patients is followed shortly after by a relapse of disease[41]. H2O2-induced inhibition of mitochondrial beta-oxidation (viamitochondrial thiolase inhibition) increases the anapleurotic metabolism of glutamine, which decreases the biosynthesis of GSH contributing to the excess colonocyte H2O2load(Figure 4). This mechanism is also consistent with a genetic predisposition that impairs the colonocyte’s ability to neutralize an H2O2load. Within this redox framework, inhibition of colonocyte beta-oxidation is a secondary effect of colonocyte xenobiotic (PFOA or bromooctanoate) metabolism, which generates excess ETC H2O2that impairs mitochondrial beta-oxidationviaH2O2induced inhibition of mitochondrial thiolase. This raises the possibility of primary prevention with an oral reducing agent for communities at risk for the adverse effects of PFOA exposure. A causal role for H2O2can be tested in the laboratory by the prevention of PFOA (or 2-bromooctanoate) induced murine colitis with an oral reducing agent (see treatment section below).
Lastly, PFOA’s adverse effects are not limited to UC. Lymphocytes are highly sensitive to the toxic effects of H2O2and undergo apoptosis at very low levels of H2O2exposure of less than 1 µm[186].Studies showing a significant association between PFOA serum levels and decreased antibody response to vaccination are consistent with this mechanism of action[182]. Thus, PFOA resistance to cellular metabolism and degradation is likely to result in excess H2O2production in any cell contaminated by this xenobiotic.
Endogenous oxidative stressors: A look inside
In UC, lifelong episodes of relapsing inflammation affecting the same colonic regions previously inflamed indicate that inflammation has fundamentally changed the colonic epithelium compared to the pre-morbid state. Since the character of the inflammation does not change over time and neutrophils continue to be the first responders streaming into the colonic epithelium, this suggests that H2O2is still the chemotactic agent involved but from a new source. The evidence points to new endogenous sources of H2O2that combine with pre-existing sources of H2O2to increase the likelihood of relapse.
Microbiome: An oxidative dysbiosis
Although disruption of the colonic microbiome can contribute to the onset of UC by decreasing butyrate production, which leads to increased colonocyte H2O2(Figure 4), UC can also adversely affect the microbiome. Studies have shown a 10 × increase in H2O2producing bacteria in biopsies of inflamed colonic tissue in individuals with UC compared to normal controls[120]. These H2O2-producing bacteria are adherent to the colonic mucosa. This suggests that chronically high levels of H2O2in the inflammatory field create an environment that selects for bacteria that produce H2O2, which are those able to tolerate the abnormally high levels of lumenal oxidative stress. Over time, this oxidative dysbiosis may replace large portions of the normal microbiome, which may not be able to survive under conditions of high H2O2-induced oxidative stress. The H2O2released by bacteria can contribute to relapse by diffusing through the epithelium to the subjacent vascular layer where it serves as a chemotactic agent to attract neutrophils into the colonic epithelium. This creates a microbiome, which is a pro-inflammatory endogenous oxidative stressor that contributes to the onset or relapse of UC by continuous H2O2production. A recent analysis examining the pathogenesis of UC concluded that “disease onset is triggered by events that alter the healthy balance of the gut microbiota, perturb the mucosal barrier, and abnormally stimulate gut immune responses”[12]. H2O2does all three.
CYP2E1 induction: A vicious cycle
As explained above CYP2E1 is an alcohol inducible enzyme that is involved in the metabolism of ethanol and other xenobiotics entering the body. H2O2is within the chain of molecular events that upregulate inducible CYP2E1[187]. This implies that chronically elevated colonocyte H2O2from any source (i.e.,ethanol or xenobiotic metabolism, oxidative stress exposure,etc) can upregulate CYP2E1. In other words, elevated colonocyte H2O2can upregulate CYP2E1 without ethanol exposure. This can cause increased sensitivity to CYP2E1 substrates, which can lead to heightened H2O2production in areas of previous inflammation when exposed to ethanol or other xenobiotics metabolized by this enzyme.Studies have shown the cells with upregulated CYP2E1 produced higher amounts of H2O2that can exit the cell[188]. This can cause increased H2O2production upon exposure to CYP2E1 substrates such as alcohol and other xenobiotics, which increases the risk of relapse orde novoUC. Under these circumstances, upregulated CYP2E1 becomes an endogenous oxidative stressor.
Mitochondrial heteroplasmy: Hard-wired for inflammation
Intracellular H2O2is normally kept within a very low picomolar range to prevent oxidative damage from this very powerful oxidizing agent[186,189]. Over time, colonocytes from individuals with UC are exposed to higher levels of H2O2due to multiple oxidative stressors including infiltrating epithelial neutrophils (mucosal inflammation), microbiome (oxidative dysbiosis), CYP2E1 induction (alcohol and xenobiotic exposure), peroxisomal beta-oxidation (high-fat diet) and ETC hyperactivity (smoking cessation),etc.The increase in colonocyte H2O2can overwhelm cellular antioxidant systems resulting in mitochondrial genetic damage[44,45]. This occurs because mtDNA is highly susceptible to H2O2-induced oxidative damage due to a lack of introns or histones, proximity to the ETC where H2O2is produced, and inefficient DNA repair mechanisms compared to nuclear DNA[37,38].
As mentioned above, H2O2-induced mitochondrial genetic damage introduces mutations into mtDNA resulting in a different genetic sequence for some of the hundreds of mitochondrial chromosomes present in a cell. The simultaneous occurrence of normal and mutated mtDNA is called mitochondrial heteroplasmy. The presence of mitochondrial heteroplasmy (mtDNA mutations) causes miscoding during the transcription of ETC proteins leading to the biosynthesis of faulty and mutated ETC subunits. The mutated ETC disrupts electron flow causing electron loss at a greater rate than normal(increased electron leakage). These electrons combine with vicinal molecular oxygen to form superoxide that is converted to H2O2by superoxide dismutase. Since the ETC and mtDNA are both in close proximity to each other within the mitochondrial matrix, any excess H2O2produced by the ETC can easily diffuse over to any one of the 2-10 chromosomes contained within a mitochondrion leading to additional mtDNA mutations and greater H2O2production[190]. This positive biofeedback mechanism establishes a vicious cycle that leads to ever-increasing levels of colonocyte H2O2.
The presence of mitochondrial heteroplasmy (and the resulting increase in colonocyte H2O2) can contribute to the tendency of UC to worsen over time with more frequent and severe episodes of relapse[7]. Relapse may occur in response to increasingly minor (oxidative) stressors due to the already high intracellular colonocyte H2O2. Thus, the accumulation of colonocyte H2O2transforms otherwise innocuous insults into ‘second hit’ stimuli. The “first hit” being the pre-existing excess of colonocyte H2O2. For example, in individuals with inactive UC, low to moderate red wine consumption (1-3 glasses daily) increased colonic epithelial paracellular permeability in areas previously affected by mucosal inflammation[123,191]. Increased colonic paracellular permeability is characteristic of individuals with UC[24,25]. And H2O2is reported to increase paracellular permeability by disrupting cellular tight junctions[20,22,192-194]. Under these circumstances, it takes less alcohol to increase colonocyte H2O2to the point where it diffuses out of the cell causing oxidative damage to tight junctions with subsequent increases in colonic paracellular permeability. This explains the second hit phenomenon in UC. In summary, mitochondrial heteroplasmy is unique because it is an ever-present self-amplifying intracellular oxidative stressor that facilitates the establishment of HCRH (Figure 2), which contributes to both the increased frequency of relapse and/or severity of disease.
Homocysteine: Inhibition of GPx
Several studies have reported significantly elevated levels of tissue and serum homocysteine in children and adults with UC[195-198]. A significant positive association between elevated homocysteine and UC was confirmed by two meta-analyses in 2011 and 2018[199,200]. Homocysteine inhibits GPx, the principal antioxidant enzyme system used by the cell to remove (neutralize) H2O2[201,202].Furthermore, homocysteine-induced inhibition of GPx occurs at physiologic levels of serum homocysteine[203]. Inhibition of GPx can increase cellular H2O2, especially during oxidative stress exposure,which acutely increases cellular H2O2production. Thus, homocysteine is a significant endogenous oxidative stressor that can increase colonocyte H2O2and contribute tode novoUC or disease relapse.
EVlDENCE-BASED TREATMENT
Therapy: Targeting H2O2
A causal role for H2O2in the pathogenesis of UC implies that induction of remission can be achieved by eliminating extracellular colonocyte H2O2while maintenance of remission is attained by normalizing intracellular levels of colonocyte H2O2. The overall objective is to abrogate the H2O2molecular chemotactic “trail” that is guiding the directed migration of subjacent intravascular neutrophils to the source of H2O2emanating from the colonic epithelium. Without an interstitial H2O2gradient signal to follow, neutrophils are no longer attracted into the colonic epithelium, which effectively terminates the inflammatory response. These were the goals that guided the formulation of a novel therapy, which consists of a topical multicomponent enema (described below) administered with a systemic oral reducing agent [R-dihydrolipoic acid (RDLA)] that targets extracellular and intracellular colonocyte H2O2respectively. RDLA is the reduced form of the biologically active ‘R’ enantiomer of lipoic acid and is the only form that should be administered as the oxidized form (described below) might worsen UC.
We administered the enema once daily (usually at bedtime) for 2 wk followed by once every other day for two weeks. Oral RDLA 300 mg twice daily was initiated when enema therapy was begun.Treatment with RDLA was continued for 4-6 mo. The components of the enema are mesalamine [5-aminosalicylic acid (5-ASA)], budesonide, sodium cromolyn, and sodium butyrate. The enema formulation and evidence-based rationale for the inclusion of each component of the entire therapy are discussed below. The severity or extension of the disease was not a consideration when initiating therapy since, in theory, all patients with UC should respond to a reduction of colonic H2O2. Our only consideration was whether the patient could tolerate the therapy.
Enema formulation
The enema was formulated by adding the following components to a standard 60-milliliter enema bottle containing 4 g of mesalamine (5-ASA) from which 20 milliliters were removed (and discarded): (1) 15 milliliters of 1 molar sodium butyrate (1.7 g); (2) 5 milliliters of sodium cromolyn (total 100 mg); and (3)1 milliliter of budesonide (5 mg/mL). Gentle swirling should follow the addition of each component to ensure uniform dispersal. The total ending volume is 61 milliliters. The combination enema is easily made by a compounding pharmacist. We only used the original enema bottle containing mesalamine to formulate this therapy as other bottles may have residual chemicals that can worsen UC.
Results of treatment
The novel treatment was offered to 36 patients with refractory UC as part of the practice of medicine(average MAYO score 8.6, range 3-12) and the results were published as a case series in which 85%achieved complete histologic remission in under 8 wk[204]. Although long-term follow-up was not part of the case series, a case report was generated after I was contacted by a patient included in this case series[205]. The patient had a 39-year history of refractory UC, which had progressed to severe pancolitis at the time of treatment when he was being considered for a total colectomy. His follow-up colonoscopy, which was performed 12 years later in 2019, showed completely normal colonic biopsies with no signs of UC. To date, (14 years after treatment) the patient relates having uninterrupted completely normal bowel movements. Based on the available data, this appears to be the first documented cure of UC. The video of an in-house clinical presentation of the first five patients to receive this new therapy with before-and-after treatment histology presented by the attending physician and attending pathologist is available[206].
Within this evidence-based redox framework, the general therapeutic intervention for all UC patients is the same regardless of duration or severity of illness, course, relapsing frequency, mucosal inflammatory distribution, age of onset, previous medications, or extra intestinal manifestations. Modifications may be required for those patients who are intolerant to any of the components in the therapy. In patients who are intolerant of topical (enema) therapy, treatment can be initiated with RDLA alone,which as an amphipathic membrane-permeable antioxidant (H2O2neutralizing) and reducing (cellular electron-donating) agent may restore colonocyte redox homeostasis and resolve mild-moderate cases of UC with more severe cases improving enough to begin topical (enema) therapy if needed.
Severe UC
Up to 25% of patients with UC will experience severe disease requiring hospitalization either on initial presentation or during the course of their illness[207]. Severe UC presenting with multiple daily episodes of bloody diarrhea and signs of systemic toxicity is a life-threatening emergency with significant morbidity, high risk of colectomy, and a pre- and post-operative mortality of up to 3% and 5% respectively[8,207,208]. In these individuals, oral and/or topical therapy may not be possible or advisable. The evolution of UC to this extreme degree indicates severe depletion of both colonic and systemic reductive capacity[61,62]. The exceptionally high rate of current medical treatment failure for severe UC and the observation that remission is associated with restoration of colonic redox homeostasis[208,209], suggests that patients with severe UC should be considered for therapy with an intravenous reducing agent such as sodium thiosulfate (STS) as part of their overall treatment regimen in order to rapidly reduce systemic and colonic H2O2, restore redox homeostasis and promote mucosal healing. Given that current management of acute severe UC is not based on high-quality evidence, the need for effective therapy is all the more pressing[210].
STS is an odorless, water-soluble, small inorganic molecule (MW-158.11 g/mol) that is normally produced in mitochondria as a product of sulfide oxidation pathways[211]. STS is on the World Health Organization’s (WHO) list of essential medicines and is supplied for intravenous use due to rapid gastric degradation[211]. STS is a direct-acting reducing agent that can donate two electrons to chemically neutralize H2O2upon contact[212]. STS will also reduce the oxidized form of GSH (GSSG)back to reduced GSH, which is needed to neutralize H2O2and maintain cellular redox homeostasis[213].The advantage of STS is that it does not depend upon biological antioxidant enzyme systems for its therapeutic action. This is beneficial in critical settings when rapid reduction of H2O2and restoration of redox homeostasis are required.
The basic chemical reaction for the reduction of H2O2with STS is: 4 H2O2+ S2O32-→ 2 SO42-+ 2 H++ 3 H2O[212]. Based on the above chemical reaction and evidence implicating a causal role for H2O2in the pathogenesis of UC, STS is expected to abrogate the interstitial neutrophilic chemotactic effect being exerted by H2O2and thus prevent neutrophil migration into the colonic epithelium, which perpetuates colonic inflammation. The reduction of extracellular colonic mucosal H2O2by STS can act as a sink that will facilitate the diffusion of intracellular colonocyte H2O2to the extracellular space where it can be neutralized by STS.
H2O2can impair smooth muscle contraction and interrupt neuromuscular transmission leading to reduced colonic muscle tone and lowered colonic lumenal pressure, which is postulated to play an essential role in the development of toxic megacolon, a life-threatening complication of UC[214-218].Thus, reducing agents such as STS or RDLA may have a role in treating or preventing toxic megacolon.Once severe UC is resolved, patients should be discharged on an oral reducing agent such as RDLA for an indefinite period of time to lower the risk of relapse.
STS is well tolerated and approved for use in cyanide poisoning with a recommended dose of 12.5 g over slow IV infusion (10-20 min) in adults and 250 mg/kg in children[219,220]. Similar dosing regimens can be considered in UC with repeat dosing guided by clinical status. STS is an accepted therapy for calciphylaxis due to chronic renal failure and is administered to mitigate the adverse effects of cisplatin toxicity during the treatment of solid tumors[213].
MAlNTENANCE: TARGETlNG OXlDATlVE STRESSORS
Although therapeutic intervention to lower H2O2and restore colonic redox homeostasis is a critical part of overall therapy, elimination of contributing lifestyle and dietary habits that increase cellular H2O2(environmental oxidative stressors, Figure 3A) must be part of the long term strategy to maintain remission. A complete list of environmental oxidative stressors and their mechanisms is beyond the scope of this paper however, some general evidence-based guidelines can be made. Stress and alcohol ingestion are major oxidative stressors and should be avoided. A low-fat diet with adequate amounts of dietary fiber, especially soluble fiber, is extremely important to minimize colonic oxidative stress[83,221-223].
Eating food that is free of pesticides, chemicals and additives is critical for maintaining a healthy microbiome. High levels of iron in red meat and some tap water are oxidative stressors and should be avoided[224,225]. Certain fish contain high levels of mercury[226,227]. Mercury is an oxidative stressor and therefore it is prudent to minimize ingestion of fish containing high levels of mercury. Studies have shown that sugar and sugar-sweetened drinks are also associated with UC[228,229]. So it is best to avoid high sugar-containing foods and drinks.
Carrageenan is a non-nutritive food additive that is used as a thickening agent in many foods.Although considered safe, food-grade carrageenan can be converted to a colitis-promoting smallmolecular (degraded) carrageenan when exposed to H2O2[230,231]. Since H2O2is present in the inflammatory field it (H2O2) can convert food-grade carrageenan to the degraded variety in the colon of individuals with UC. Studies report that degraded carrageenan can penetrate colonocytes and generate superoxide, which is converted to H2O2[232]. Thus, carrageenan is an oxidative stressor that generates intracellular colonic H2O2and should avoid by individuals with UC.
Smoking cessation is a strong oxidative stressor and should be undertaken very slowly in patients with UC to avoid relapse. RDLA can be administered during smoking cessation to counteract oxidative stress. Patients should be very cautious with vitamin supplementation because studies have shown that certain vitamins such as vitamin B6 (pyridoxine) have been associated with the development of UC[233]. Pyridoxine used in supplements and food fortification is converted to the biologically active pyridoxal by pyridoxine 4-oxidase (EC1.1.3.12), which produces H2O2as a by-product[234]. Adequate sleep (at least 6 h) and regular moderate exercise are very important for individuals with UC to reduce stress[235,236]. Based on the data indicating a compromised reductive capacity, all UC patients should be maintained on an oral reducing agent (i.e.,RDLA) for an indefinite period of time. The following section provides a detailed explanation regarding the scientific basis for each component of the therapy.
Rationale for multicomponent enema therapy
As mentioned above, the compound enema contains four components: Mesalamine (5-ASA),budesonide, sodium cromolyn, and sodium butyrate. The therapeutic rationale for the compound enema is based on the mechanism of action for each component to act in an additive fashion in order to decrease colonic H2O2as indicated below. The base component, 5-ASA has an anti-inflammatory effect that is limited to the specific type of inflammation present in UC and does not have a positive therapeutic action on colonic inflammation in general[237]. This suggests that 5-ASA’s mechanism of action is directed at the causal agent responsible for the inflammation in UC. This is in contrast to other currently available therapeutic agents used to treat UC, which have a more general immunosuppressive action in the colon. 5-ASA’s mechanism of action is that of a topically-acting extracellular tetra-valent reducing agent capable of donating up to 4 electrons per molecule for the reduction(neutralization) of H2O2and other oxygen radials[238,239]. Since 5-ASA is able to induce and maintain histologic remission in active UC, this indicates that 5-ASA can neutralize extracellular neutrophilderived H2O2in the inflammatory field leading to induction of remission while the sustained topical epithelial presence of 5-ASA maintains remission by neutralizing H2O2diffusing from colonocytes.
Neutralization of colonocyte-derived H2O2by 5-ASA prevents the establishment of an H2O2mediated neutrophilic chemotactic gradient, which attracts neutrophils into the colonic epithelium leading to relapse. This is supported by the observation that 5-ASA’s histologic remission rate of nearly 45% is the highest of any currently available therapeutic approved to treat UC suggesting 5-ASA’s ability to neutralize colonic H2O2is interrupting a fundamental underlying biological process (i.e.,neutrophilic chemotaxis) in the molecular chain of events leading to UC[240].
Butyrate, a short-chain fatty acid normally produced by the colonic microbiome as a colonocyte fuel source, is the second component. Butyrate reduces the anapleurotic metabolism of glutamine, which increases colonocyte GSH. This augments the colonocyte’s capacity to neutralize intracellular H2O2(Figure 4). Studies have shown significantly increased colonic epithelial GSH after topical butyrate administration, and high fiber diets that generate increased fecal butyrate are reported to maintain prolonged remission in patients with UC[104,241].
The third component, cromolyn sodium, is a mast cell stabilizer that inhibits the secretion of histamine by mast cells, which accumulate in large amounts at sites of tissue injury in UC[242-244].Mast cells are significantly activated in UC and undergo intense degranulation resulting in the secretion of significantly greater amounts of histamine that is concentrated at sites of colonic inflammation in UC[245-248]. Rectal biopsies of patients with UC contained significantly higher amounts of histamine compared to normal controls[249,250]. Histamine is degraded by diamine oxidase (EC 1.4.3.22) whose activity is especially high in the intestinal mucosa and the inflammatory field with H2O2being the product of this reaction[245,251,252]. Thus, cromolyn prevents histamine from being secreted from mucosal mast cells, which precludes its conversion to H2O2that can significantly contribute to the intensity and persistence of colonic inflammation in UC. This is consistent with studies reporting that histamine drives the severity of inflammation in a murine model of experimental UC[253].
Budesonide is the fourth component and is a topically acting corticosteroid that inhibits neutrophil infiltration into the colon by down-regulating neutrophil and endothelial surface adhesion molecules,which prevents neutrophil attachment to the endothelium and subsequent directed migration into the colonic epithelium[254]. Neutrophils produce a large amount of H2O2viasurface NADPH oxidase[255].Corticosteroids reduce the expression of neutrophil surface NADPH oxidase thus decreasing neutrophil production of H2O2[254]. The combination of inhibited neutrophilic epithelial migration and decreased production of H2O2significantly reduces this large source of H2O2, which is a significant driving factor of mucosal inflammation in UC.
RATlONALE FOR RDLA
Targeting H2O2 with RDLA
RDLA, the oral component of the therapy, is the biologically active (dextrorotatory) reduced enantiomer of alpha-lipoic acid (the oxidized form)[256]. Alpha-lipoic acid is synthesized in mitochondria and plays an essential role as a co-factor for several multi-enzymatic complexes involved in mitochondrial energy metabolism[257]. Alpha-lipoic acid is enzymatically reduced to RDLA (the reduced form)viadihydrolipoamide dehydrogenase (E.C. 1.8.1.4) in mitochondria. Thus, alpha-lipoic acid acts as an oxidizing agent that may worsen colonocyte redox homeostasis and should not be administered to patients with UC (Figure 5).
Aside from its enzymatic role in energy metabolism, RDLA is a powerful biological reducing(antioxidant) agent that can be administered orally[263]. RDLA’s dithiol group can donate electrons to reduce (reactivate) oxidized forms of other cellular antioxidants such as vitamin-C, vitamin-E, and GSH[264]. RDLA is both a water and lipid-soluble (amphipathic) molecule so it is deliveredviathe bloodstream to all cells of the body where it diffuses through cell membranes to deliver needed reducing equivalents for the reduction of H2O2and synthesis of GSH[265]. Studies in mice demonstrate that the recycling of GSH is critical for cell survival when exposed to oxidative stress (i.e.,H2O2)[266].Other studies show that GSH protects rat intestinal epithelial cells from H2O2-induced injury[267].RDLA’s capacity to directly react with H2O2combined with its ability to significantly increase cellular GSH, the principal cellular H2O2reducing agent, underlie RDLA’s ability to combat the high levels of colonocyte H2O2in UC.
RDLA significantly increases nuclear factor E2-related factor 2, a master antioxidant transcription factor that mediates the expression of antioxidant genes, including those for GSH synthesis[268]. This significantly increases the cellular capacity to synthesize GSH and neutralize H2O2. RDLA is reported to reduce nuclear transcription factor-kappa B and adhesion molecule expression[268], which downregulates the inflammatory response and decreases neutrophilic infiltration into the colonic epithelium contributing to the resolution of inflammation and colitis. Thus, RDLA prevents colonocyte cell death during periods of oxidative stress (H2O2exposure) and promotesde novosynthesis and recycling of GSH in order to keep cellular GSH high and H2O2low. Hence, RDLA’s mechanism of action indicates that it can significantly contribute to inducing and achieving remission in UC.
Studies have shown that restoration of depleted mitochondrial GSH can reverse oxidant (i.e.,H2O2)induced mtDNA damage, which leads to mitochondrial heteroplasmy[46]. Since RDLA is highly effective at increasing cellular GSH, this suggests that RDLA will be also effective at reversing H2O2induced mtDNA oxidative damage and subsequent mitochondrial heteroplasmy that is postulated to contribute to life-long relapse. This is supported by the continuous 14-year biopsy-proven histologic remission in a patient with a 39-year history of severe refractory UC after treatment with a regimen containing RDLA[205].
RDLA is generally considered safe and is approved for the treatment of diabetic peripheral neuropathy in Europe[269]. Oral lipoic acid at doses as high as 1800 mg/d for 6 mo and 1200 mg/d for 2 years did not result in serious adverse effects when used to treat diabetic peripheral neuropathy[270,271].Studies indicate that 40% of RDLA is quickly absorbed systemically after oral dosing and rapidly distributed to tissues[263]. RDLA undergoes renal excretion and intracellular beta-oxidation[268,272],which provides a second pathway to increase cellular GSH by decreasing the anapleurotic metabolism of glutamate (Figure 4).
RDLA is the only amphipathic orally administered therapeutic that is both an intracellular and extracellular anti-oxidant (H2O2neutralizing) and reducing agent (electron-donating for maintenance of redox homeostasis). Given these highly unique and desirable therapeutic properties, which are essential for long-term remission in UC, RDLA should be made widely available and be included on the WHO’s list of essential medications. STS, an intravenous reducing agent, is already on the WHO’s list of essential medications and thus RDLA, a therapeutically active oral reducing agent should also be included. In addition to UC, emerging evidence suggests that RDLA (and STS) may have a preventive and/or therapeutic role in other diseases in which the evidence indicates a causal role for H2O2such as systemic lupus erythematosus, sepsis, and diabetes[273-275].

Figure 5 R-dihydrolipoic acid. A: R-dihydrolipoic acid (RDLA) is the reduced form of alpha lipoic acid (the oxidized form); B: The reducing equivalents of RDLA are provided by its two thiol groups (red circles) that are each capable of donating one electron. RDLA has a redox potential of -290 millivolts which is only exceeded by NADH and NADPH with a redox potential of -320 and -400 millivolts respectively[258]. Due to its very low (more negative) redox potential, RDLA can directly or indirectly reduce all other cellular antioxidants and many types of oxygen radicals[256]. These include vitamin-C, vitamin-E, glutathione, thioredoxin, glutaredoxin,catalase, glutathione peroxidase, and peroxiredoxin[258-261]. Alpha-lipoic acid is reduced by dehydrolipoamide dehydrogenase and Thioredoxin reductase, which use NADH and NADPH as reducing co-factors respectively[262]. The amphipathic nature of RDLA (lipid and water-soluble) allows it to diffuse throughout cellular compartments to transport reducing equivalents where needed and assist in neutralizing excess hydrogen peroxide. The elimination of excess intracellular cellular hydrogen peroxide is essential in order to restore cellular redox homeostasis and prevent ulcerative colitis relapse caused by extracellular diffusion of colonocyte hydrogen peroxide. Vit: Vitamin; DLD: Dehydrolipoamide dehydrogenase; TRx: Thioredoxin reductase; GSH: Glutathione.
In summary, an evidence-based analysis of the pathogenesis and therapy of UC indicates that treatment of inflammation is no longer the main objective of this illness. Instead, the primary goal is the restoration of colonic and systemic redox homeostasis by therapeutic normalization of colonic H2O2,which removes the molecular chemotactic signal that initiates and maintains colonic inflammation and is responsible for disease relapse.
DlSCUSSlON
The existence of an unpredictable, unexplainable, and incurable disease such as UC indicates that we are guided by the wrong theory of pathogenesis in our quest to develop effective treatment and find causes and cures. Currently, there are two, mutually exclusive, mechanisms of disease that have been put forward to explain how UC develops and guide therapeutic development. Both mechanisms attempt to answer the same question; why do white blood cells (neutrophils) suddenly leave the surrounding blood vessels and head straight into the epithelial lining of the large intestine causing inflammation, bleeding, and UC?
The first hypothesis is consensus-based, agreed upon among researchers in the field, and posits an immune abnormality as a primary event in the development of UC while the second mechanism is evidence-based and maintains that the immune system is completely normal but only appears to be“attacking” the colon due to the inappropriate secretion of a neutrophilic chemoattractant, H2O2, by the colonic epithelium, which draws neutrophils into the epithelial lining. Despite decades of intensive research, no evidence of any antecedent immune abnormality has ever been identified in individuals with UC or their immediate relatives, and studies of basic immune functionality in UC patients are normal[10,11]. Due to the absence of a biologically plausible mechanism, the term “immune dysregulation” has been coined to explain the presence of neutrophils in the colonic epithelium. In contrast,studies have demonstrated significantly increased H2O2production in non-inflamed colonic epithelium prior to the appearance of mucosal inflammation, satisfying the absolute chronological requirement that the cause (H2O2) must precede the effect (inflammation). Since H2O2is cell membrane permeable, once it leaves the colonocyte H2O2can establish a chemotactic gradient “trail” that neutrophils can follow right into the colonic epithelium after exiting the subjacent vasculature. Hence, within this framework and in line with previous data, the immune system in UC is normal. Neutrophils are just doing what they are biologically programmed to do when exposed to H2O2; a normal chemotactic immune signaling agent being inappropriately secreted by the colonic epithelium.
On a clinical level, immune dysregulation cannot explain any of the basic characteristics that define UC such as why people develop this disease to begin with, what is the genetic predisposition, why it always starts in the rectum, what causes proximal inflammatory progression, why the loss of intestinal hemostasis leading to bloody diarrhea, what is the mechanism behind relapse, what causes colonic epithelial crypt abscesses, why are smoking cessation, low fiber and high-fat diets risk factors for UC,what dietary and lifestyle changes will help prevent relapse, what therapeutic intervention will provide long-lasting remission and how can we effectuate a cure, to name a few. In other words, immune dysregulation, as a mechanism of disease, has no explanatory power, which is essential for understanding a disease process. In comparison, a redox mechanism of disease based on colonocyte buildup of H2O2explains these basic observations and provides a consistent and clear mechanistic foundation to understand the hereto-forth puzzling observations that characterize UC.
Despite the absence of any hard evidence for an immune abnormality, this hypothesis continues to be the main focus of ongoing investigation by “leading researchers” in the field who have issued consensus statements asserting that a “dysregulated immune response” is the “widely accepted” cause of UC[276,277]. A consequence of leading researcher support for immune dysregulation is the near-total focus of therapeutic development on proprietary and costly drugs that alter the immune response in a limited number of commercially viable but non-curative ways[6]. As a result, induction trials for new drug development have reached an unsurpassable therapeutic ceiling of 20%-30% indicating that treatment aimed solely at modifying the immune response cannot alter the natural history of the disease, which is essential for achieving universal long-lasting remission in patients with UC[278]. In other words,immune dysregulation as a mechanism of disease has no predictive power to identify a discreet causal agent, which can be targeted for effective treatment and curative potential.
In contrast, the evidence-based identification of H2O2as a causal agent in UC has guided the development of highly effective treatment with at least one documented, biopsy-proven, histologic remission lasting 14 years to date without any episodes of intervening relapse, in a patient with a 39-year history of refractory UC[205]. This is the basis of bench-to-bedside translational medicine that integrates basic science discoveries into predictable effective treatments and potential cures. A causal therapeutic target eliminates much of the “trial and error” that defines the history of current UC therapeutic development[279].
Nevertheless, what leading researchers think is highly relevant for patients with UC. A consensus mechanism of disease put forth and agreed upon among researchers means that clinicians do not have an evidence-based pathogenesis to guide clinical decision-making. This forces clinicians to rely on a variety of clinical variables including the severity of disease, colonic inflammatory distribution, age of onset, previous medication, disease duration, disease course, relapse frequency, and extra-intestinal manifestations to make bedside patient care determinations[7]. Since each one of these variables can be different for every patient, the number of treatment guidelines must be numerous enough to encompass all these different patient combinations. These treatment guidelines, in turn, are not founded on evidence that defines the pathogenesis but instead rely on an ever-growing and changing database of empirical studies incorporating one or more of these myriad clinical variables. The interpretation of these clinical studies is consensus-driven by committee and thus inherently subjective, leading to numerous clinical recommendations for UC that range in number from 32 to 124 treatment guidelines depending on the country of origin[2].
Committee members may also disagree about the relevance of any particular study for the treatment of UC leading to differences in the number of treatment guidelines between medical societies in the same country with the American Gastroenterological Association espousing 24 treatment recommendations while the American College of Gastroenterology supports 49 clinical treatment guidelines for the treatment of this highly debilitating inflammatory bowel disease[280-282]. Moreover, due to the“inconsistencies regarding recommendations” between the two societies, a “Guide to Guidelines in UC”was published in an effort to reconcile the differences among leading clinicians in the field[283]. In contrast, an evidence-based H2O2mechanism of disease only has one guideline for the treatment of UC that does not change, which is to normalize colonic H2O2.
The current degree of therapeutic uncertainty when treating patients with UC is the inevitable result of not having an evidence-based mechanism of disease as the foundation for clinical decision-making and therapeutic development. In other words, since there is no evidence for an antecedent immune vulnerability in UC, treatment with the sole aim of suppressing the immune response is not anchored in an evidence-based pathogenesis. The end result is subjective and malleable clinical guidelines with shifting therapeutic targets generating different empirical treatments, the majority of which are based on low or very low-quality evidence while being permeated by high degrees of conflict of interest[2].Patients ultimately bear the brunt of these fluid upstream decisions because treatments based on lowquality evidence cannot alter the natural history of disease leading to a high degree of medical treatment failure and a 30% colectomy rate[5]. The high degree of medical treatment failure, in turn, fuels endless fund-raising to pay for research in order to find a more effective therapy. And research, unfortunately,continues to be guided by the same consensus immune dysregulation hypothesis ultimately degenerating into a perpetual sisyphean iterative endeavor.
Perhaps the most relevant outcome when applying the predictive power of an H2O2evidence-based mechanism of disease to UC is the expectation of indefinite remission and normal colon functionality once excess colonic H2O2is neutralized. Given a causal role for H2O2in UC, the elimination of excess colonic H2O2would abrogate the molecular signal for directed neutrophil migration into the colonic epithelium leading to long-term histologic (and biochemical) remission. Accordingly, colonic inflammation is not the principal focus of treatment, instead, the primary therapeutic goal is to remove the H2O2mediated chemotactic signal attracting neutrophils into the colonic epithelium.
This represents a functional cure as long as intracellular colonocyte H2O2remains normal and unable to diffuse into the extracellular microenvironment. Treatment limited to reducing inflammation does not address H2O2emanating from colonocytes, and thus cannot cure. The continued build-up of colonocyte H2O2while being treated with these drugs can increase mitochondrial heteroplasmy with worsening disease and/or lead to colon cancer due to the genotoxic effects of H2O2[50,51].
From a redox medicine perspective, inflammation (neutrophil infiltration) is just one source of H2O2that must be addressed. Other sources contributing H2O2to the colonic inflammatory field in UC such as mast cells (histamine), EC (serotonin), and microbiome oxidative dysbiosis must also be considered for optimal therapeutic intervention to induce remission. Environmental oxidative stressors and mitochondrial heteroplasmy, which channel H2O2viathe colonocyte into the colonic epithelium have a crucial role in provoking relapse and must be addressed in order to achieve long-lasting remission.
Additionally, the common metabolic origin of cellular H2O2suggests that H2O2-mediated intestinal inflammation is not solely confined to the colon. A recent analysis concluded that a shared mechanism underlies UC and UC-associated ileitis, which develops in up to 35% of patients with UC[284]. This is supported by studies in UC patients showing impaired ileal butyrate oxidation in both terminal ileum and colon, which in the latter was associated with H2O2induced inhibition of mitochondrial thiolase, the last enzyme in the butyrate beta-oxidation cascade[30,285]. This suggests that excess H2O2is responsible for impaired ileal butyrate oxidation in the small intestine as well. Moreover, the neutrophilic epithelial infiltration, cryptitis, and crypt abscesses that characterize UC-associated ileitis is analogous to the typical histopathological changes observed in UC[286]. This strongly implies that H2O2is also elevated in the small intestine leading to mucosal inflammation and metabolic derangements. Thus, treatments that simply target colonic inflammation do not address the consequences of elevated ileal H2O2, which may lead to small bowel inflammation and interfere with the absorption of nutrients. In contrast,treatment with a systemic reducing agent such as RDLA has the potential of resolving UC-associated ileitis.
The interdisciplinary nature of evidence-based analysis can provide clues to understanding and effectively treating other serious conditions that are linked to UC whose medical therapy has so far been suboptimal. Studies have shown that H2O2can effectively inhibit neuromuscular transmission[216].Protection against H2O2-induced inhibition of neuromuscular transmission was associated with the cellular ability to eliminate H2O2[287]. Other studies have demonstrated that H2O2contributes to motor disfunction in human UC[218]. This suggests a potential causal role for H2O2in the motility dysfunction that is thought to underlie toxic mega colon and small bowel bacterial overgrowth, both of which are associated with symptomatic UC[214,288]. It also implies that therapeutic intervention with reducing agents (STS or RDLA) to reduce colonic H2O2may be an effective therapeutic option in treating or preventing these serious conditions.
At the other end of the clinical spectrum, up to 2% of asymptomatic individuals undergoing screening colonoscopy were shown to have typical histologic inflammatory features of UC with twothirds developing symptomatic disease (rectal bleeding) within 5 years[289,290]. Despite having a high probability of developing symptomatic UC and the possibility of increased risk of colon cancer,clinicians are in a quandary regarding the appropriate treatment for these asymptomatic individuals since all medications used to treat UC can have serious side effects and there are no data regarding their effectiveness at this very early asymptomatic stage[291-293]. However, the presence of preclinical neutrophilic inflammation implies that H2O2has begun “leaking” out of colonocytes and is attracting neutrophils into the colonic epithelium. The use of an oral reducing agent (RDLA) to normalize colonocyte H2O2and restore redox homeostasis would be a logical choice at this stage due to its recognized safety profile and ability to enhance cellular reductive capacity in order to reduce colonic H2O2. If future studies show that this is an effective treatment, it will be possible to prevent symptomatic disease from developing while UC is still in a preclinical asymptomatic stage.
All things considered, with current therapy and under the best of circumstances, UC patients must undergo life-long surveillance colonoscopy for colon cancer, which cannot detect all neoplastic lesions leading to a high mortality rate[29,294,295]. And although total colectomy has been touted as a cure for the approximately 30% of UC patients who fail medical therapy or develop colon cancer, studies have shown that patients who have undergone ileal pouch/anal anastomosis have higher disability scores than patients with active UC[296]. In contrast, maintenance therapy with an oral reducing agent has the potential of eliminating the need for colectomy, lifelong colonoscopies, and, by removing excess colonic H2O2, may significantly reduce the incidence of UC-associated colorectal cancer.
An evidence-based theory of UC identifying H2O2as a causal therapeutic target not only has the potential of highly effective and inexpensive treatment that may be curative but opens the door to population-wide primary prevention by increasing total reducing equivalents (antioxidants) in our food supply. This is supported by studies demonstrating a decreased risk of developing UC with diets high in anti-oxidants (reducing agents)[297]. Dietary intervention can be successful in reducing the incidence of UC because, in contrast to established disease with high levels of colonic H2O2that require treatment with powerful reducing agents, intervention before colonocytes develop HCRH (while intracellular H2O2is still low) requires much less reductive capacity, which can be supplied by increasing the amount of reducing equivalents (antioxidants) in the food supply. This public health level intervention may prevent UC from developing in the entire population before it even starts.
CONCLUSlON
The evidence supports a causal role for colonocyte H2O2in the pathogenesis and pathophysiology of UC. Treatment to reduce and maintain normal colonic H2O2levels leads to long-term histologic remission (complete mucosal healing) in patients with refractory disease. The treatment is inexpensive and well tolerated. Lifestyle modifications to reduce oxidative stress exposure will help maintain remission. This is the first time that a causal evidence-based therapeutic target with curative potential has been identified for UC. The inclusion of multiple components to address the different sources of H2O2within the colitic inflammatory field contributes to its singular effectiveness but also slows its acceptance by a healthcare system dominated by single molecular therapeutics.
H2O2is a normal by-product of cellular metabolism that can accumulate within colonic epithelial cells. H2O2’s unique properties of cell membrane permeability, long life, potent oxidizing potential, and the ability to attract white blood cells combine to promote colonocyte extracellular diffusion followed by oxidative disintegration of colonic epithelial tight junctional proteins while simultaneously attracting white blood cells into the colonic epithelium, both of which lead to colonic inflammation and eventual UC. This makes it appear as if the immune system is “attacking” the colon when in reality the immune response is behaving as it is normally programmed to respond. The abnormality is the inappropriate secretion of H2O2by the colonic epithelium and not the immune response.
The pleiotropic effects of H2O2have misdirected the careers of multiple generations of researchers into searching for a non-existent primary immune abnormality as the cause of UC. Extensive research since the mid-20thcentury has failed to uncover a primary immune vulnerability to explicate this illness.Cumulative evidence does not support any form of immune dysregulation in the pathogenesis of UC.This line of research is not evidence-based and should be abandoned. The continued search for immune dysregulation as the cause of UC leads to enormous research waste and endless fundraising that will never find the cause or cure for this disease while at the same time encouraging the treatment of UC with expensive immune altering agents that drive up healthcare costs, do not cure and are associated with lower quality of life, higher rates of colon cancer and other serious adverse effects. Continued research to uncover a primary immune abnormality as the cause of UC reinforces a false sense of hope for millions of individuals suffering from this illness who are desperately waiting for a cure that will never materialize with this line of research. Only by following the evidence can we cure disease.
Evidence-based medical research will eventually displace consensus-driven hypothesis in the highly competitive race for research funding as the National Institute of General Medical Sciences begins to shift funding priorities to grant applications that can clearly explicate falsifiable disease mechanisms that are “associated with the pathogenesis and resolution” of disease[298]. This pathogenesis initiative has begun with sepsis in July 2019 and is likely to be expanded as a requirement to obtain scarce research funding for other diseases as well. Under these guidelines, the current consensus-based immune dysregulation hypothesis invoked to explain UC does not meet this threshold for Federal research funding since it neither provides a coherent falsifiable pathogenesis nor a means of disease resolution.
A causal role for colonic H2O2in the pathogenesis of UC is biologically plausible and supported by both experimental and clinical evidence. H2O2satisfies all the basic requirements for an etiological agent leading to the development of UC and is worthy of continued and expanded research to confirm a potential causal role in the pathogenesis of this debilitating inflammatory bowel disease affecting millions worldwide. It is incumbent upon the research community to follow up on this highly promising line of research that raises the real possibility of targeted and highly effective treatment with curative potential.
Future directions
Complex diseases such as UC arise as an emergent systems property of its individual,in vivo, interacting constituent elements. The physical proximity of the colonic epithelium, innate immune system, and colonic lumenal contents gives rise to the disease phenotype we call UC in response to colonocyte extracellular diffusion of H2O2. Complex chronic diseases such as UC are not amenable to a reductionist analytical laboratory approach that examines each contributing element outside of its disease-producingin vivocontext[299]. The prevalence of “incurable” complex chronic diseases continues to rise because they have slipped through the cracks of our current reductionist methodology of medical research that is not designed to detect emergent systems diseases such as UC. The shortcoming of laboratory research to deal with chronic complex diseases can be overcome with graduate programs dedicated to theoretical(systems) medicine, which looks at the big picture to help guide laboratory researchers down a focused experimental pathway to discovering causes and cures of disease[300]. With chronic disease mortality accounting for 60% of all global deaths as well as 70% of all deaths in the United States, and 60% of Americans suffering from at least one chronic disease with 40% afflicted with two or more chronic ailments, chronic disease has become the leading driver of the United States’ $3.5 trillion in annual health care cost[301-303]. We simply cannot win the war against encroaching chronic disease by experimentation alone. This underscores the critical need for collaboration between systems medicine(theoretical systems pathogenesis) and laboratory-based experimentalists (reductionist medical research) before the financial, emotional, and familial burden becomes too much to bear and society begins to destabilize under the weight of too many sick people.
FOOTNOTES
Author contributions:Pravda J is the sole author of this manuscript and solely responsible for its content; Pravda J performed all the research, collected, analyzed, and interpreted all the data; Pravda J conceived of and developed the hydrogen peroxide-based pathogenesis of ulcerative colitis; Pravda J prepared and wrote the manuscript and performed all critical revisions; Pravda J certifies that this manuscript is the product of his original research; and Pravda J has overall responsibility for this manuscript.
Conflict-of-interest statement:The author reported no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORClD number:Jay Pravda 0000-0001-5737-5506.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Gastroenterology2022年31期
World Journal of Gastroenterology2022年31期
- World Journal of Gastroenterology的其它文章
- Duodenal-jejunal bypass reduces serum ceramides via inhibiting intestinal bile acid-farnesoid X receptor pathway
- Preoperative contrast-enhanced computed tomography-based radiomics model for overall survival prediction in hepatocellular carcinoma
- Prevalence and clinical characteristics of autoimmune liver disease in hospitalized patients with cirrhosis and acute decompensation in China
- Application of computed tomography-based radiomics in differential diagnosis of adenocarcinoma and squamous cell carcinoma at the esophagogastric junction
- Radiomics and nomogram of magnetic resonance imaging for preoperative prediction of microvascular invasion in small hepatocellular carcinoma
- Insights into induction of the immune response by the hepatitis B vaccine