
HP-RTM 高活性树脂体系制备及性能
2022-08-17 09:15张欢陈杰王美发郭东朋
工程塑料应用 2022年8期
张欢,陈杰,王美发,郭东朋
(深圳市飞荣达科技股份有限公司,广东深圳 518132)
高压-树脂传递模塑成型(HP-RTM)是指通过高压压力将树脂对冲混合并注入到预先铺设有纤维增强材料和预置嵌件的真空密闭模具内,经树脂流动充模、浸渍、固化和脱模,获得复合材料制品的成型工艺,是近年来推出的一种应对大批量生产高性能热固性复合材料零件的新型工艺技术,主要应用于新能源汽车、航空航天等轻量化领域[1–2]。相比传统的纤维复合材料成型工艺需要消耗大量的人力和时间,HP-RTM 可实现低成本、短周期(大批量)、高质量生产,保证了纤维的快速浸润和优异的产品性能[3–4]。
但HP-RTM 工艺对树脂基材的要求很高,不仅要求黏度低、凝胶时间适当长、固化速度快,还要求阻燃性能好、耐热性好以及力学性能高等。目前,HP-RTM 工艺用树脂制备技术主要掌握在欧美等国家化工巨头手中,已经形成了较成熟的产业化,其中代表性的有美国亨茨曼公司、陶氏公司、美国瀚森公司、美国巴斯夫公司等。国外化工巨头凭借其技术优势,高价出售其树脂,同时从国外进口还存在运费高、仓储和保质不便利等缺点。
然而国内市场没有能同时满足上述要求的商业树脂,普遍存在凝胶时间短或固化速度慢、阻燃效果差或耐热性能低等问题,高性能树脂还处于研究阶段。国内有些单位也开发了类似的树脂配方体系,但仍处于研究阶段。刘钟铃等[5]针对HP-RTM工艺用快速固化环氧树脂(EP)体系,研究了其固化反应动力学及流变行为,研究成果为HP-RTM 成型工艺参数设定与优化提供了技术基础和理论依据。王佳明等[6]就树脂传递模塑成型的材料、工艺、设备、应用等方面进行了总结,并预测了其未来的发展方向,对树脂传递模塑工艺未来的研究有一定的借鉴性。朱怡臻等[7]概述并分析了传统树脂传递模塑(RTM)成型工艺过程及优缺点,并对HP-RTM,轻质树脂传递模塑成型、真空辅助树脂灌注工艺、树脂联合注射浸渍技术等RTM 衍生成型工艺的研究进展与应用进行了介绍分析。于德润等[8]介绍了RTM 成型工艺原理、RTM 衍生工艺成型过程及特点、RTM 工艺应用现状以及RTM 用树脂的研究现状,并提出了RTM 工艺进一步发展亟待解决的问题。满足HP-RTM 工艺的树脂仍然严重依赖进口。因此,研发一款满足国内市场需求的HP-RTM 高活性树脂具有重要的意义与价值。
笔者采用EP 为基体,1,4-丁二醇二缩水甘油醚为稀释剂,十溴二苯乙烷、聚磷酸铵为阻燃剂,聚丙二醇二缩水甘油醚为增韧剂,2-乙基-4-甲基咪唑为促进剂,多乙烯多胺为固化剂,制备出了HPRTM 高活性树脂体系。研究了稀释剂用量对高活性树脂体系的黏度、凝胶时间、固化速度、阻燃性能、热性能、力学性能和储能模量的影响。
1 实验部分
1.1 主要原材料
EP:E-51,廊坊诺尔信化工有限公司;
1,4-丁二醇二缩水甘油醚:Syna-Epoxy 27,南通新纳希新材料有限公司;
十溴二苯乙烷:含量≥99.8%,济南金盈泰化工有限公司;
聚磷酸铵:标准料,济南金盈泰化工有限公司;
2-乙基-4-甲基咪唑:E104846,上海阿拉丁生化科技股份有限公司;
聚丙二醇二缩水甘油醚:EPG-217,环氧值0.11~0.16 ep/100 g,烟台奥利福化工有限公司;
多乙烯多胺:P108302,上海阿拉丁生化科技股份有限公司。
1.2 主要设备与仪器
电子天平:BN-V8-600 型,厦门巨林仪器有限公司;
磁力搅拌水浴锅:SHJ-4AB 型,常州金坛良友仪器有限公司;
模块化智能型高级流变仪:MCR 102 型,奥地利安东帕(中国)有限公司;
精密鼓风干燥箱:KJ-2010A 型,东莞市科建检测仪器有限公司;
水平-垂直燃烧测试仪:AUTO-SP 型,深圳奥德赛创科技有限公司;
动态热机械分析(DMA)仪:Q800 型,美国TA仪器公司;
数显式简支梁冲击试验机:XCJD-50 型,承德市金建检测仪器有限公司;
场发射扫描电子显微镜(FESEM):S4800 型,日本HITACHI 公司。
1.3 试样制备
HP-RTM 高活性树脂体系A 组分制备:将100质量份的EP,1~5 份2-乙基-4-甲基咪唑,15~20份聚丙二醇二缩水甘油醚,以及1,4-丁二醇二缩水甘油醚以一定比例混合,于60~80℃反应1 h,混合搅拌均匀后,得到A 组分,冷却至室温待用。其中1,4-丁二醇二缩水甘油醚的质量分数分别为0,5%,10%,15%,20%。
B 组分制备:将100 份多乙烯多胺,20~30 份十溴二苯乙烷,10~15 份聚磷酸铵,于室温下反应0.5 h,混合搅拌均匀后,得到B 组分待用。
固化体系制备:将A 组分和B 组分在室温下以100 ∶20 质量比混合均匀,将混合后的树脂体系浇注到预先涂有脱模剂的模具中,抽真空脱除气泡30 min,再以100℃/3 min 进行快速固化,最后自然冷却至室温脱模,制得测试样品。
1.4 性能测试与表征
黏度按照ISO 3219–1993 测试,测试A 组分黏度,温度30~100℃;
凝胶时间按照GB/T 12007.7–1989 测试,测试A 组分与B 组分混合后的凝胶时间,温度设置25~100℃;
固化速度测试是将A 组分与B 组分混合后,放入100℃烘箱中,记录其完全固化的时间;
垂直燃烧性能按照UL 94 测试,样条厚度1 mm;
玻璃化转变温度(Tg)按照ASTM D7028–2007测试,样条尺寸15 mm×12 mm×4 mm,温度25~200℃;
储能模量按照ASTM E2254–2009 测试,温度25~200℃;
冲击强度按照GB/T 1043.1–2008 测试,冲击能量5.5 J,速度3.8 m/s,跨距62 mm;
用FESEM 观察样品的冲击断面,经喷金处理,15 kV 电压,放大200 倍。
2 结果与讨论
2.1 稀释剂用量对树脂体系A 组分黏度的影响
树脂体系的黏度特性是表征HP-RTM 工艺参数的一个重要指标,黏度低有利于树脂对纤维的渗透和浸润,而加入稀释剂是降低树脂黏度最有效的方法[9]。同时根据设备生产经验,黏度过高会导致注射树脂时喷口压力剧增,使得A,B 组分配比失调,注射口附近有不能完全固化情况,从而产生不良品。因此,加工工艺要求A 组分的黏度30℃时小于15 Pa·s,100℃时小于0.1 Pa·s。
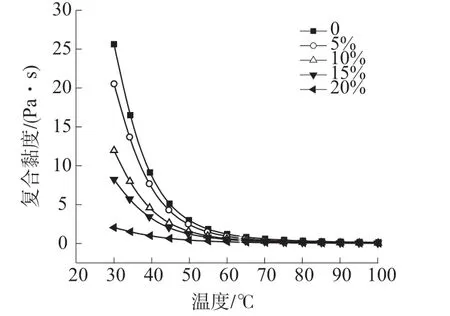
图1 稀释剂不同用量的树脂体系A 组分黏度
图1 是从室温到工作温度范围内,稀释剂1,4-丁二醇二缩水甘油醚不同用量的HP-RTM 高活性树脂体系A 组分的黏度,表1 是对应的黏度具体数值。由图1 可以看出,随着温度的升高,树脂体系的黏度快速下降,达到70℃以后黏度下降幅度很小。由表1 可以看出,与不含稀释剂的树脂比较,当稀释剂质量分数为5%时,室温下黏度下降并不明显,说明加入质量分数为5%时较少,稀释作用还未体现,不足以满足工业生产要求;当稀释剂质量分数超过10%以后,稀释作用开始体现,树脂黏度下降明显,特别是当其质量分数为20%时,黏度进一步快速下降。说明在此HP-RTM 高活性树脂体系配方中,1,4-丁二醇二缩水甘油醚的质量分数要大于10%,但从成本上考虑不能超过20%,否则不具备竞争优势。

表1 稀释剂不同用量下树脂体系A 组分黏度 Pa·s
2.2 稀释剂用量对树脂体系A,B 组分凝胶时间的影响
对于HP-RTM 工艺生产的大尺寸复杂结构复合材料产品,通常要求树脂的凝胶时间要适当长,这样在注射时就有较长的适应期,树脂流动覆盖范围大。根据注射时间的要求,该HP-RTM 高活性树脂体系的凝胶时间最佳范围是70~100 s。
图2 是在稀释剂1,4-丁二醇二缩水甘油醚的不同用量下,A,B 组分混合后温度随时间的变化规律。图3 是A,B 组分混合后复合黏度随时间的变化规律。表2 是A,B 组分混合后具体凝胶时间。

图2 稀释剂不同用量下A,B 组分混合后温度随时间的变化

图3 稀释剂不同用量下A,B 组分混合后复合黏度随时间的变化
由图2 可看出,A,B 组分混合后加热到工作温度100℃的时间基本一致,约200 s,说明稀释剂对树脂体系升温速率基本没有影响。从图3 可看出,随着稀释剂用量的增加,A,B 组分混合后复合黏度升高所需要的时间在逐渐增加,体现在表2 中就是A,B 组分混合后的凝胶时间在延长。说明稀释剂能延长HP-RTM 高活性树脂体系凝胶时间,且用量越多作用越明显。但凝胶时间也不能过长,否则影响树脂体系的固化速度,导致生产效率下降,这点下文分析。所以在该树脂体系中稀释剂的最佳质量分数为10%和15%。

表2 稀释剂不同用量下A,B 组分混合后凝胶时间
2.3 稀释剂用量对树脂体系A,B 混合组分固化速度的影响
在HP-RTM 工艺中要求树脂的固化速度快,以提高生产效率,实现短周期大批量生产。该HPRTM 高活性树脂体系技术指标要求5 min 以内快速固化。图4 是在稀释剂的不同用量下,AB 组分在100℃条件下固化后的照片。

图4 稀释剂不同用量下A,B 组分固化后的照片
由图4 可以看出,随着稀释剂用量的增加,A,B 混合组分固化后的表面由粗糙逐渐变得光滑,且没有气泡。表3 是A,B 混合组分的固化情况。由表3 可以看出,随着稀释剂用量的增加,A,B 组分固化时间变长。这是因为,一方面稀释剂延长了HP-RTM 高活性树脂体系的凝胶时间;另一方面,稀释剂的稀释作用有利于固化反应产生的大量热及时散发出去,以免热量促进固化反应。而当不加稀释剂或只加少量5%时,虽然有更快的固化速度,但在固化过程中产生大量黑烟,边缘聚集大量气泡。这也是因为固化速度太快,体系大量放热,热量来不及耗散,使未固化小分子氧化冒烟,从而也使固化后的表面变得粗糙有气泡。

表3 稀释剂不同用量下A,B 组分混合后的固化情况
2.4 稀释剂用量对树脂体系阻燃性能的影响
该HP-RTM 高活性树脂体系固化后要求阻燃性能达到UL94 V–0 级,所以增加了20~30 份十溴二苯乙烷和10~15 份聚磷酸铵阻燃剂。表4 是稀释剂不同用量下固化体系阻燃性能的测试结果。由表4 可以看出,当不加稀释剂或加入量小于等于15%时,此HP-RTM 高活性树脂体系都能达到UL94 V–0 级;而当质量分数增加到20%后,虽然余焰和余燃时间都达标,但有滴落物点燃棉花,从而使阻燃等级降为UL 94 V–2。这也是因为1,4-丁二醇二缩水甘油醚的稀释作用使树脂流动性增加,燃烧后导致滴落,进一步说明1,4-丁二醇二缩水甘油醚用量不能太大。
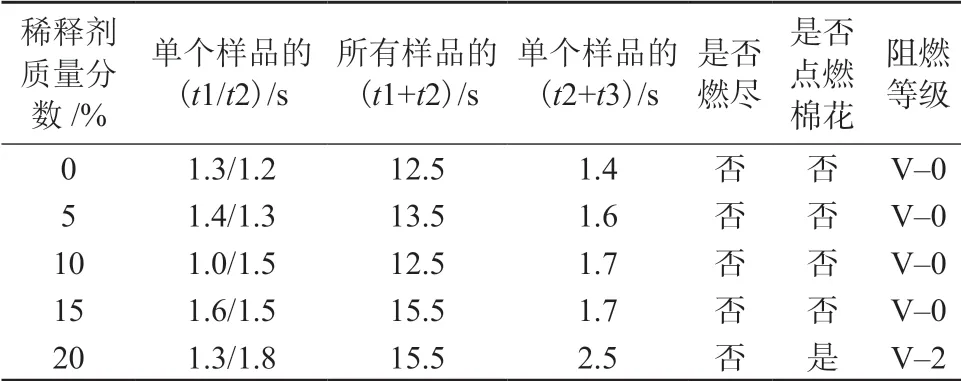
表4 稀释剂不同用量下固化体系的阻燃情况
2.5 稀释剂用量对树脂体系热性能的影响
因HP-RTM 高活性树脂体系应用在新能源汽车电池包上,对耐热性能有一定要求,技术指标要求Tg≥110℃。图5 是稀释剂不同用量的HP-RTM 高活性树脂体系的DMA 曲线。由图5 可以看出,随着稀释剂1,4-丁二醇二缩水甘油醚用量的增加,树脂体系的Tg逐渐降低,当稀释剂的质量分数为15%时HP-RTM 高活性树脂的Tg是113.72℃,此时仍满足要求,当稀释剂的质量分数为20%时HP-RTM高活性树脂的Tg是101.10℃,已不能满足实际应用使用要求。这是因为相较于基体EP,1,4-丁二醇二缩水甘油醚的分子量较小,当加入1,4-丁二醇二缩水甘油醚后树脂体系的分子量会降低。而对于同一种聚合物,在一定的分子量范围内,其耐热性与分子量大小成正比,分子量降低,Tg下降[10–11]。
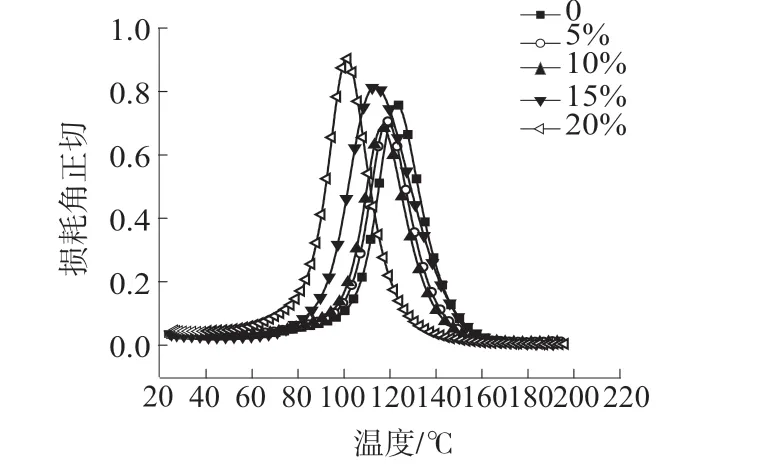
图5 稀释剂不同用量下的HP-RTM 高活性树脂体系的DMA 曲线
2.6 稀释剂用量对树脂体系储能模量的影响
图6 是稀释剂不同用量的HP-RTM 高活性树脂体系的储能模量。由图6 可以看出,随着稀释剂用量的增加,树脂体系的储能模量逐渐降低。在工作温度100℃下,当稀释剂质量分数为15%时HPRTM 高活性树脂体系储能模量是485.8 MPa,此时仍满足要求,但当稀释剂质量分数为20%时HPRTM 高活性树脂体系储能模量是112.3 MPa,此时材料偏软,难以满足脱模要求。这也是因为稀释剂降低了树脂体系的分子量,从而使储能模量下降。
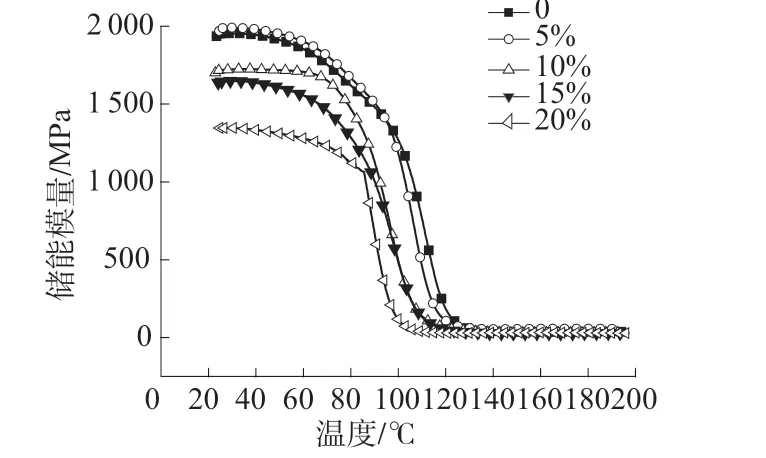
图6 稀释剂不同用量下的HP-RTM 高活性树脂体系的储能模量
2.7 稀释剂用量对树脂体系冲击强度的影响
图7 是不同稀释剂用量的HP-RTM 高活性树脂体系的冲击强度。由图7 可以看出,随着稀释剂用量的增加,HP-RTM 高活性树脂体系的冲击强度呈现先增大后减小的趋势,当1,4-丁二醇二缩水甘油醚质量分数为10%时HP-RTM 高活性树脂体系的冲击强度达到最大值15.3 kJ/m2,当1,4-丁二醇二缩水甘油醚质量分数为15%时体系的冲击强度为12.5 kJ/m2。这是因为,一方面与EP 基体相比,稀释剂分子结构中有可挠性长链,可以自由旋转而富有弹性,引入到HP-RTM 高活性树脂体系中后,降低交联密度,提高交联网络活动能力,使得HPRTM 高活性树脂体系韧性提高[12–13];另一方面,随着稀释剂用量的增加,A,B 组分固化后的表面由粗糙逐渐变得光滑,且没有气泡,从而提高HP-RTM高活性树脂体系的冲击强度。但当稀释剂用量过多时,HP-RTM 高活性树脂体系的分子量快速下降,从而使各项性能下降。说明在一定范围内稀释剂的加入有利于提高HP-RTM 高活性树脂体系的冲击强度。
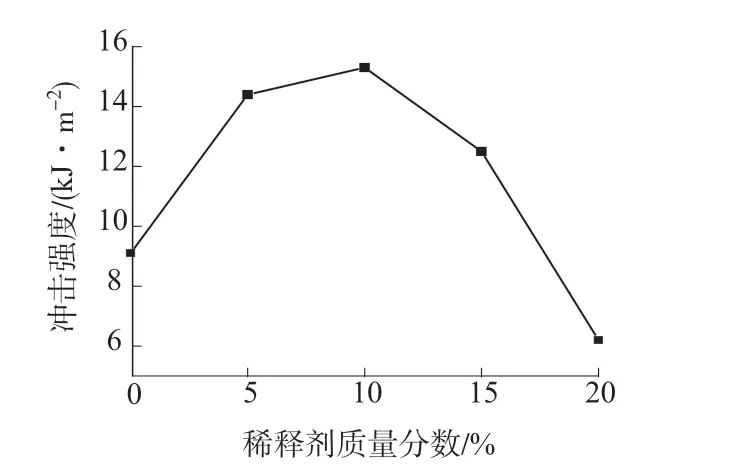
图7 稀释剂不同用量的HP-RTM 高活性树脂体系的冲击强度
图8 是稀释剂质量分数为0 和15%时HPRTM 高活性树脂体系的冲击断面的SEM 照片。由图8 可以看出,当不加稀释剂或加入质量分数为15%时,断裂面粗糙且高低错落,裂纹出现明显的分支且较密,大量微裂纹在发展过程中受到抑制而终止,阻止了裂纹的进一步扩展,吸收了部分冲击能,呈现韧性断裂[14–15]。这是因为该HP-RTM 高活性树脂体系中增加了15~20 份聚丙二醇二缩水甘油醚增韧剂,但加入质量分数15%的稀释剂后,韧性断裂趋势更为明显,所以HP-RTM 高活性树脂体系的冲击强度提高。
3 结论
制备了1,4-丁二醇二缩水甘油醚稀释剂质量分数分别为0,5%,10%,15%,20%的HP-RTM 高活性树脂体系。结果表明,在一定范围内,1,4-丁二醇二缩水甘油醚稀释剂可以降低HP-RTM 高活性树脂体系的黏度、固化速度,但延长凝胶时间会降低HP-RTM 高活性树脂体系的阻燃性能、玻璃化转变温度、储能模量,提高了冲击强度。综合考虑,当1,4-丁二醇二缩水甘油醚稀释剂质量分数为15%时,HP-RTM 高活性树脂体系的性能最优,30℃时黏度是8.237 Pa·s,100℃时是0.044 Pa·s,凝胶时间是96.4 s,固化时间是5 min,且固化过程中不产生黑烟,固化后表面变得光滑没有气泡,垂直燃烧性能达到UL94 V–0 级,玻璃化转变温度是113.72℃,储能模量是485.8 MPa,冲击强度达到12.5 kJ/m2。此高活性树脂体系满足实际生产和应用要求。
猜你喜欢
广州化工(2022年11期)2022-06-29
科学家(2022年3期)2022-04-11
粘接(2021年5期)2021-06-29
森林工程(2020年5期)2020-09-17
核化学与放射化学(2020年4期)2020-08-21
石油研究(2019年7期)2019-09-10
天然产物研究与开发(2019年1期)2019-03-01
科学与技术(2018年8期)2018-04-26
报刊荟萃(上)(2018年2期)2018-03-03
中国化工贸易·上旬刊(2017年4期)2017-09-10
