
基于序列深度学习的III型分泌效应子预测
2022-08-16 03:11唐贤俊王顺芳
计算机工程与设计 2022年8期
唐贤俊,王顺芳
(云南大学 信息学院,云南 昆明 650504)
0 引 言
III型分泌效应子(T3SE)被认为是许多宿主-微生物相互作用的主要介质[1,2]。T3SE作为宿主内的毒力因子[3],能够改变和操纵重要的宿主细胞功能[4]。由于最近菌株的不断扩大和基因组学数据的可用性持续增加为深入研究提供了独特机会[5,6],因此对T3SE的预测具有重要的研究意义。迄今为止,已经有相当多的T3SEs通过体外和硅胶方法鉴定[7]和基于经典的机器学习计算方法,例如支持向量机(SVM)[8]、Naive Bayes(NB)[9]、卷积神经网络(CNN)[10]和马尔可夫模型(MM)[11]。
机器学习方法需花费大量时间提取有意义的特征来进行预测,它们无法直接从一级序列中直接进行预测。并且这些预测方法存在大量潜在的假阳性,因此将具有高度预测精度的基于序列深度学习的预测方法应用于III型分泌效应子具有挑战性。鉴于此,本文提出了基于深度学习的方法从一级序列预测III型分泌效应子。该方法跳过了从序列对象中提取特征的步骤,能够利用两级卷积神经网络自动学习特征,用双向长短时记忆神经网络识别长期依赖关系,最后用二进制交叉熵来评价神经网络质量。最终在数据集上的实验结果表明,该方法预测能力强、通用性强、可靠性高。同时,该方法大大节省了预测时间的开销。
1 深度学习模型结构
就像现实世界中的自然语言一样,字母以不同的组合形式组合在一起构成单词,单词以不同的方式组合在一起形成短语。对文档中的词进行处理可以传达出文档的主题及其有意义的内容。在这项工作中,蛋白质序列类似于文档,氨基酸类似于单词。挖掘它们之间的关系将产生与序列对应的物理实体的行为属性的更高层次的信息。
我们这里所提出的深度学习模型由5个层次组成:编码层、嵌入层、CNN层、BiLSTM层和Dense层,如图1所示。
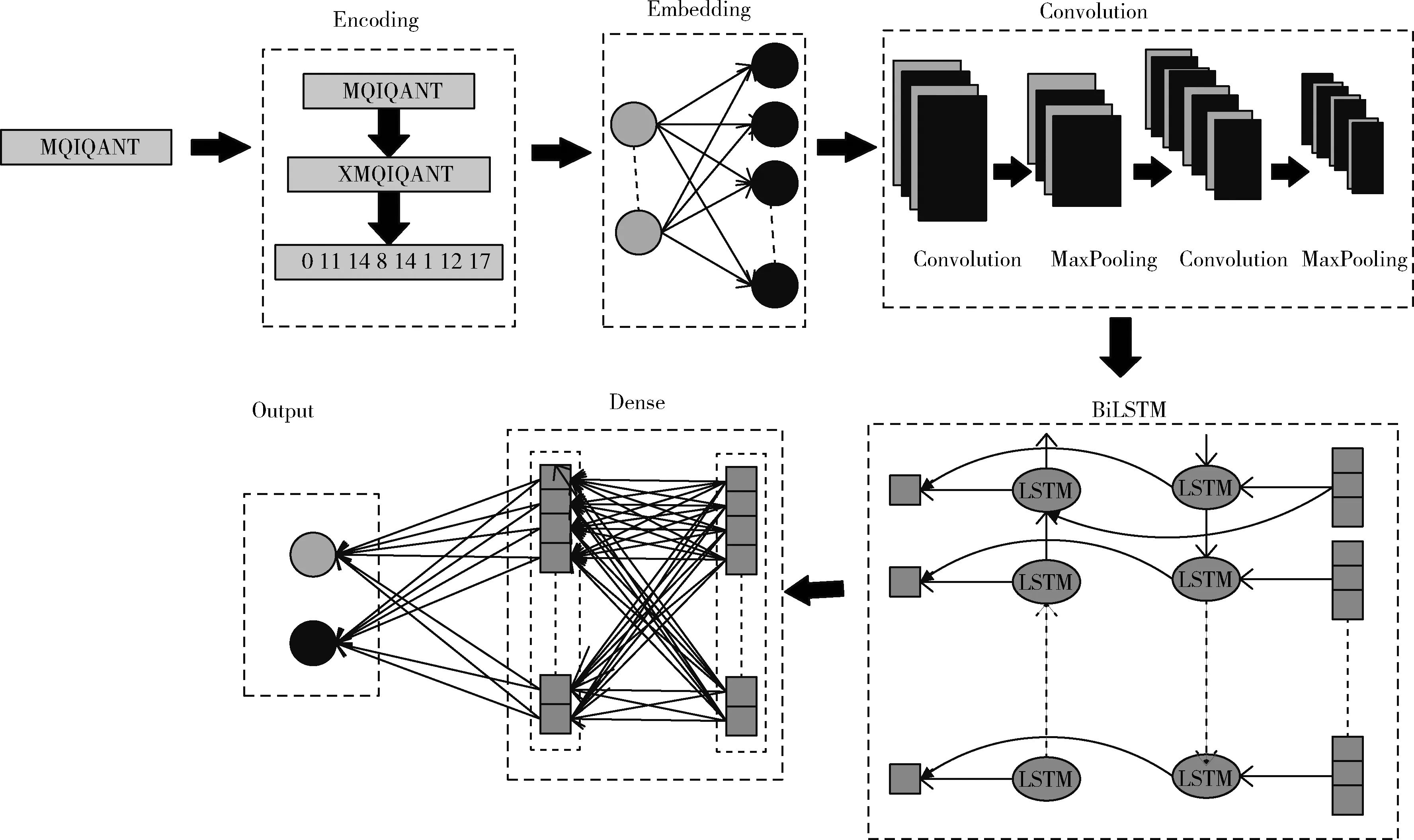
图1 深度学习模型结构
编码层主要是将序列映射到固定长度的数字向量。嵌入层的功能是将其转换为连续向量。与word2vec模型[12]类似,转换到这个连续空间可以使用连续度量的相似性概念来评估单个氨基酸的语义质量。CNN层则是由两个卷积层和两个最大池化层构成,其中每个卷积层后面都有一个最大池操作。CNN可以在多层神经元之间实施局部区域连接模式,以利用空间上的局部结构特征。具体地说,CNN层用于捕捉蛋白质序列的非线性特征,并增强与DNA结合功能的高级关联。BiLSTM层用来学习基序之间长期依赖关系。Dense层用来进行维度变换,把BiLSTM的输出转换成需要的维度输出。最后,用sigmoid激活来预测蛋白质序列的功能标签。给定一个蛋白质序列,最后我们可以用式(1)进行描述
G(S)=Dense(BiLSTM(CNN(Embedding(Encoding(S)))))
(1)
1.1 蛋白质序列编码
在大多数分类任务中,特征编码是建立统计机器学习模型的一项繁琐而关键的工作。人们提出了多种方法,如基于同源性的方法、基于PSSM的方法和基于理化性质的提取方法等。虽然这些方法在大多数情况下都能很好地工作,但人们的高度参与导致实际应用中的用处较小。在新兴的深度学习技术中,最成功的是它能够自动学习特征。为了验证其通用性,我们只给每个氨基酸分配一个自然数,见表1。

表1 氨基酸编码
编码阶段只是生成一个蛋白质序列的固定长度的数字载体。如果出现蛋白质序列的长度小于预设定的max_length值,则在前面填充一个特殊标记“X”。我们使用一个示例序列S=MQIQANT来进行表示,若我们把max_length设置为8,则它在编码时的序列填充为:S=XMQIQANT, 最后编码的值如式(2)所示
S1=Encoding(S)=(0,11,14,8,14,1,12,17)
(2)
1.2 嵌入阶段
在自然语言处理中,向量空间模型被用来表示单词。嵌入是一个映射过程,将离散词汇表中的每个单词嵌入到一个连续的向量空间中。这样,语义上相似的词被映射到相似的区域。这种映射关系在反向传播的过程中,是一直在更新的,因此能在多次epoch后,使得这个关系变成相对成熟,即:正确的表达整个语义以及各个语句之间的关系.现有的深度学习开发工具包Keras里就提供了一个嵌入层,可以将代表词汇表中每个单词的整数维矩阵进行转换。因此在嵌入层之后,输入的氨基酸序列变为密集的实值向量序列。
1.3 卷积阶段
卷积神经网络改善了传统模式识别方法存在提取特征难的问题,不仅具有传统神经网络的优点,并且还具有自动提取特征、权值共享的特点,因而在图像处理中得到了广泛的应用。编码的氨基酸序列在通过嵌入层时被转换成一个固定大小的二维矩阵,因此可以像图像一样通过卷积神经网络进行处理。激活函数使用的是“Relu”,旨在提高网络的非线性特性。最大池层通过下采样策略降低前一层输出的维数。并且我们测试了2~6范围内的各种池大小以获得最佳性能。然后,选择了池长长度为2的最大池池化操作,以最大值作为过滤器得到对应的特征。
1.4 BiLSTM阶段
虽然传统的RNN在语音识别和文本生成方面取得了显著的成果,但是梯度的消失和爆炸问题使得学习长期动态变得困难。BiLSTM是由LSTM组合而成,LSTM详细定义参见文献[13]。使用BiLSTM,我们能够在指定的时间范围内有效地使用过去的特征(通过前向状态)和未来的特征(通过后向的状态)。我们使用通过时间的反向传播(BPTT)来训练双向LSTM网络。想要了解BiLSTM,首先我们要明白LSTM模型的工作原理。LSTM模型的具体工作原理可参见文献[13]。
1.5 Dense层
全连接层(fully connected layers,FC)在整个卷积神经网络中起到“分类器”的作用。全连接层可以整合卷积层或者池化层中具有类别区分性的局部性。全连接层的具体描述请参见文献[14]。
1.6 激活和损失函数
一般来说,sigmoid函数表现出良好的数学行为。它是一个可微的有界函数,在各点均有非负的导数。当x趋于正无穷大时,f(x)趋近于1;当x趋于负无穷时f(x)趋近于0。常用于二元分类(binary classification)问题,以及神经网络的激活函数(activation function)(把线性的输入转换为非线性的输出)。所以在这项工作中,用sigmoid函数作为网络的激活函数。
损失函数衡量机器学习模型与经验数据的拟合程度。它是一个非负实值函数,损失函数越小,模型的鲁棒性就越好。在本文中,我们使用的二元交叉熵,如式(3)。其中n是所有训练数据的数目,y是真实的标签,ypred是预测的标签
(3)
整个实验过程是在Keras2.2.4框架下实现的,Keras框架是一个极简和高度模块化的神经网络库。Keras的开发重点是支持快速实验,并支持CPU和GPU。Keras是用Python编写的,能够在TensorFlow或Theano之上运行。
2 实验结果及分析
2.1 数据集
Dillon,M.M.等[15]最近通过分析多个丁香科种复合菌株和组合多个公共序列数据库,从丁香科植物全基因组中鉴定出T3SEs。在通过使用CD-HIT[16]来去除相似冗余序列后,最终得到617个T3SEs,我们将这617个T3SEs作为正样本。我们从Uniprot数据库(2020.05)中选择多个蛋白质序列形成的负样本数据集,由于非T3SEs蛋白的数量远大于正样本的数量,为克服正、负数据集之间的不平衡现象,将这个负样本集经过和正样本相同的处理后,最终得到了616个非T3SEs作为负样本。因此最后得到的丁香型假单胞菌III型分泌效应蛋白数据集,里面包含了617个T3SEs和616个非T3SEs。我们利用这个数据集开发了基于深度学习的预测工具。
此外,为了验证我们提出模型的通用性,还使用了Li等[7]在论文中使用的两个测试集(革兰氏阴性细菌III型分泌蛋白数据集和独立测试集)。革兰氏阴性细菌III型分泌蛋白数据集[7]是通过广泛的文献检索,从中获得I型到VIII型的所有分泌蛋白的信息,并从Swiss-Prot和TrEMBL下载了相应的序列(UniProt,2008)整合而得到的。此外,还收集了Wang等和Tay等实验证实的T3SEs来提高T3SE数据的综合性[7]。革兰氏阴性细菌III型分泌蛋白数据集[7]通过使用序列标识截止值为30%的CD-HIT[13,16]来去除相似序列,然后进一步删除蛋白质序列小于100的序列。最后,得到了一个包含283个T3SEs和312个非T3SEs蛋白的革兰氏阴性细菌III型分泌蛋白数据集。独立测试集是通过检索CD-HIT法和BLAST法对整个训练数据集的成对识别率分别为小于60%和25~60%的蛋白质,从而制备了这个数据集。独立测试集测试样本包括了35个T3SEs和86个非T3SEs。在我们的整个实验过程中,所用到的数据集具体见表2。
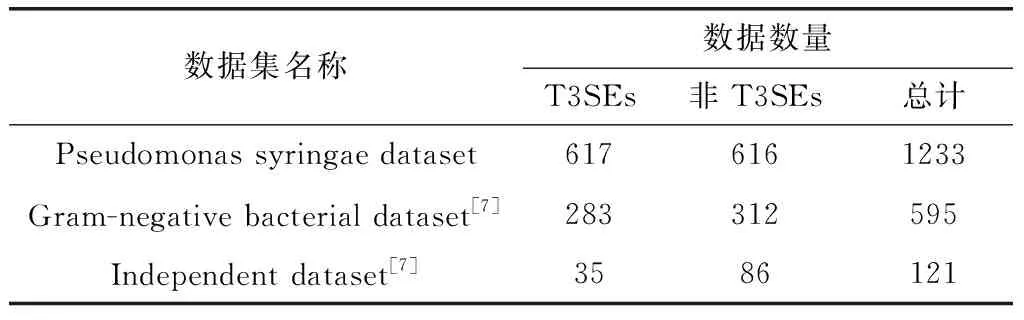
表2 数据集
2.2 性能评估指标
交叉验证是评估分类模型性能的常用方法。在这项研究中,我们将数据集分成5个不重叠的大小相等的集合,并在对其余集合进行测试。这样重复5次,5个测试集中的每一个都被用作测试集一次,并记录平均性能参数。对于二类分类问题,采用精确率(PRE)、特异性(SP)、灵敏度(SN)、准确率(ACC)、F值(F-score)和Matthew相关系数(MCC)6个参数来评价分类模型的整体预测性能。此外,还绘制了工作特性(ROC)曲线,即真阳性率与假阳性率的曲线图,以直观地衡量不同方法的综合性能。曲线下的面积(AUC)也在每个ROC图中表示了出来。AUC的最大值为1.0,表示一个完美的预测。随机猜测的AUC值为0.5。精确率(PRE)、特异性(SP)和灵敏度(SN)参数的描述可以参见文献[7],准确率(ACC)、F值(F-score)和Matthew相关系数(MCC)定义如式(4)、式(5)、式(6),其中TP指原来是正样本,分类成正样本的数量。FP指本来是负样本,却被分类成正样本的数量。TN指原来是负样本,分类成负样本的数量。FN指本来是正样本,却被错误分类成负样本的数量
(4)
(5)
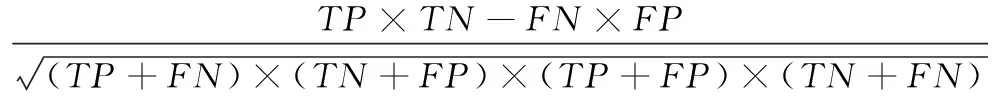
(6)
2.3 序列分析
我们分析了丁香型假单胞菌III型分泌效应蛋白每个位置上的氨基酸发生情况(包括那些代表过多和不足的氨基酸)。图2是617个T3SE和616个非T3SE效应子的50个N端和C端的位置特异性氨基酸序列图谱(图A是T3SE效应子的N末端和C末端的位置特异性氨基酸序列,图B是非T3SE效应子的N末端和C末端的位置特异性氨基酸序列)。
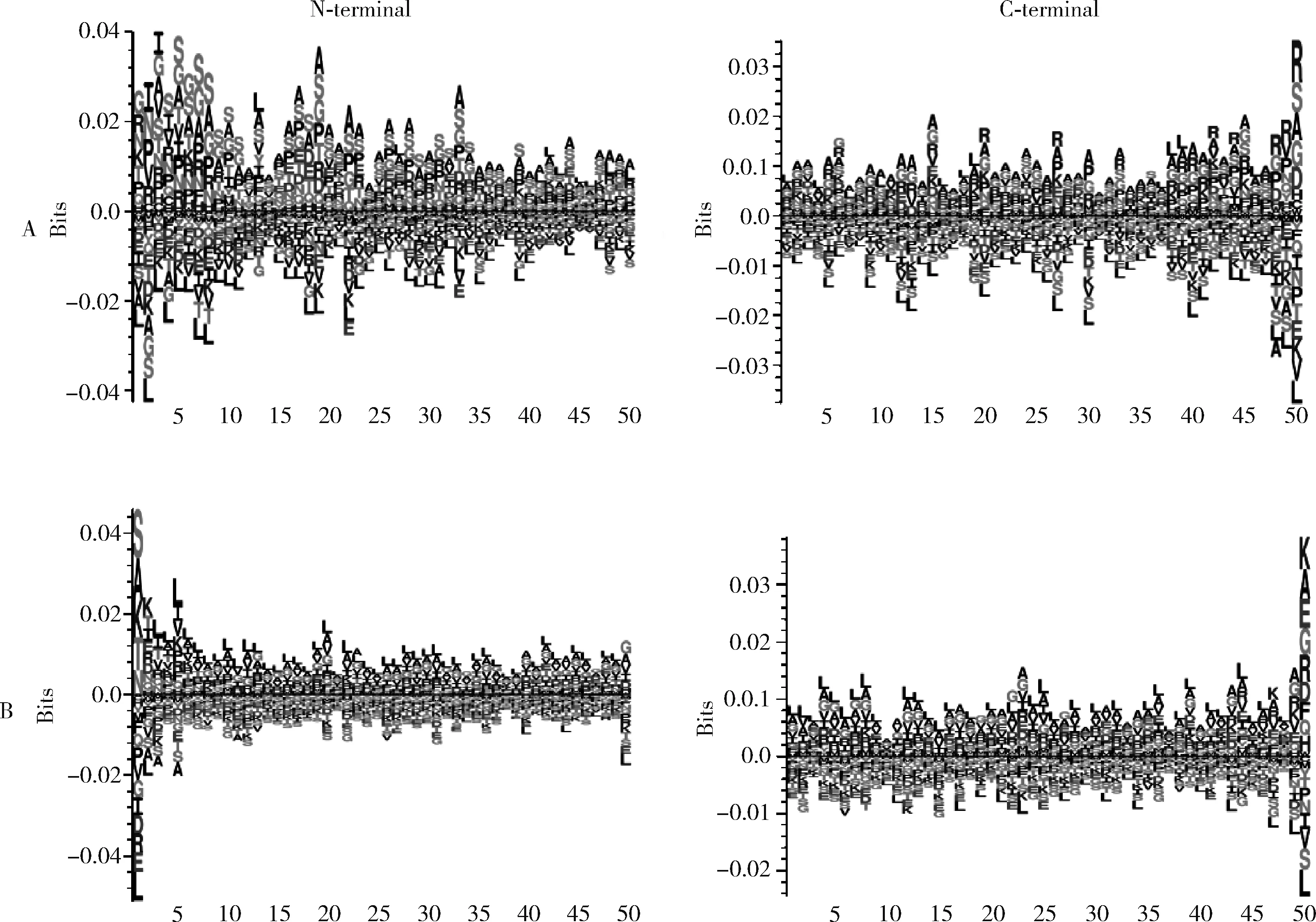
图2 位置特异性氨基酸序列分析
该图像是使用Seq2Logo默认设置生成的。y轴的正数按位表示氨基酸的丰富信息量,而y轴的负数表示相应的消耗氨基酸。横轴表示N 或C端的位置编号。对于每个序列负责翻译起始位置的第1位甲硫氨酸(M)被去除以提高可读性。在此,堆叠的高度表示每个位置的保守性水平,而字母的大小表示每个氨基酸的相对频率。
具体描述如图2所示。在图2中观察到了几个明显的氨基酸残基偏好。对于N端,在T3SE序列的多个位置上均富含丙氨酸(A)、甘氨酸(G)、丝氨酸(S),在位置2和3上富含异亮氨酸(I)。在非T3SE序列的多个位置上均富含丙氨酸(A)、亮氨酸(L),在位置1、2和5上富含赖氨酸(K),位置1上富含丝氨酸(S)。对于C端而言,在T3SE序列的多个位置上均富含丙氨酸(A)、精氨酸(R),在位置1、4、16、18、19、37、38、39和46上富含亮氨酸(L)。在非T3SE序列的多个位置上均富含亮氨酸(L),在位置10、11、17、21、23、30、32、33、43和49上富含丙氨酸(A),位置50上富含赖氨酸(K)。
所计算的氨基酸频率没有显示出在蛋白质的任一末端具有高度保守的序列基序的迹象。实际上,在C末端序列的位置50处发现了唯一可识别的具有高保守水平的位(是第二高堆栈的两倍)。但是,对于非效应蛋白也发现了相似的高保守性。T3SE的N端显示出明显均匀的保守分布,表明这些位置均未在识别中起主要作用。在T3SE的C端序列中没有明显的基序模式。基于上述的分析能将T3SE与非T3SE效应子更有效的区分开来,对于在深度学习模型中捕获蛋白质序列特征很有用。
2.4 基于五折交叉验证的分类器性能分析
为了验证该方法预测丁香型假单胞菌III型分泌效应蛋白的能力,我们在丁香型假单胞菌III型分泌效应蛋白数据集上通过k折(k=3,5,10)交叉验证实验,最终在使用5折交叉验证实验中获得了最佳模型。因此我们选择了5折交叉验证来进行接下来的比较。通过对数据集的5折交叉验证测试,我们的模型取得了良好的性能。精确率(PRE)、敏感度(SN)、特异性(SP)、F值(F-score)、准确率(ACC)、马修相关系数(MCC)、AUC的值分别为0.945、0.677、0.958、0.789、0.952、0.658、0.944。
评估分类器精度的方法我们使用了ROC图,这是分析分类器整体性能的常用方法。它将真阳性率描述为假阳性率的函数,在敏感性和特异性之间进行不同的权衡。AUC通常被用作诊断准确性的一种总结性测量。然后,将我们的模型与SVM、RF、DT、ANN、KNN和NB模型的预测结果进行了比较(图3)。我们的模型在精确率(PRE)、特异性(SP)、F值(F-score)、准确率(ACC)、马修相关系数(MCC)、AUC方面优于其它模型。我们模型的敏感度(SN)只有0.677,略低于DT模型和KNN模型的0.709和0.686。
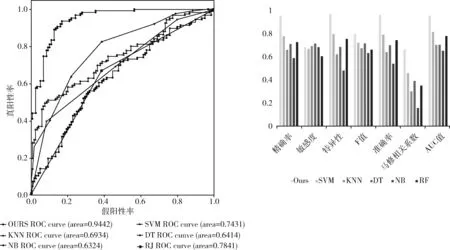
图3 基于五折交叉验证的分类器性能分析
2.5 在革兰氏阴性细菌上与其它模型的比较
首先使用283个T3SEs和313个非T3SEs的数据集训练模型。为了衡量我们提出的模型预测T3SEs的能力,我们采用了基于5折交叉验证的测试策略。将我们的模型与CNN、SVM、RF、ANN、KNN和NB模型的预测结果进行了比较。我们使用同一个革兰氏阴性细菌III型分泌蛋白数据集作为输入来训练所有模型。然后使用基于5折交叉验证测试这个数据集。
我们模型的精确率(PRE)、敏感度(SN)、特异性(SP)、F值(F-score)、准确率(ACC)、马修相关系数(MCC)、AUC分别为0.915、0.768、0.937、0.835、0.857、0.719、0.926。其中我们模型的精确率(PRE)、特异性(SP)、F值(F-score)、准确率(ACC)、马修相关系数(MCC)、AUC都优于CNN、SVM、RF、ANN、KNN和NB模型。只有敏感度(SN)略低于NB模型和CNN模型的敏感度(SN)。不同模型得到的性能具体描述见表3。
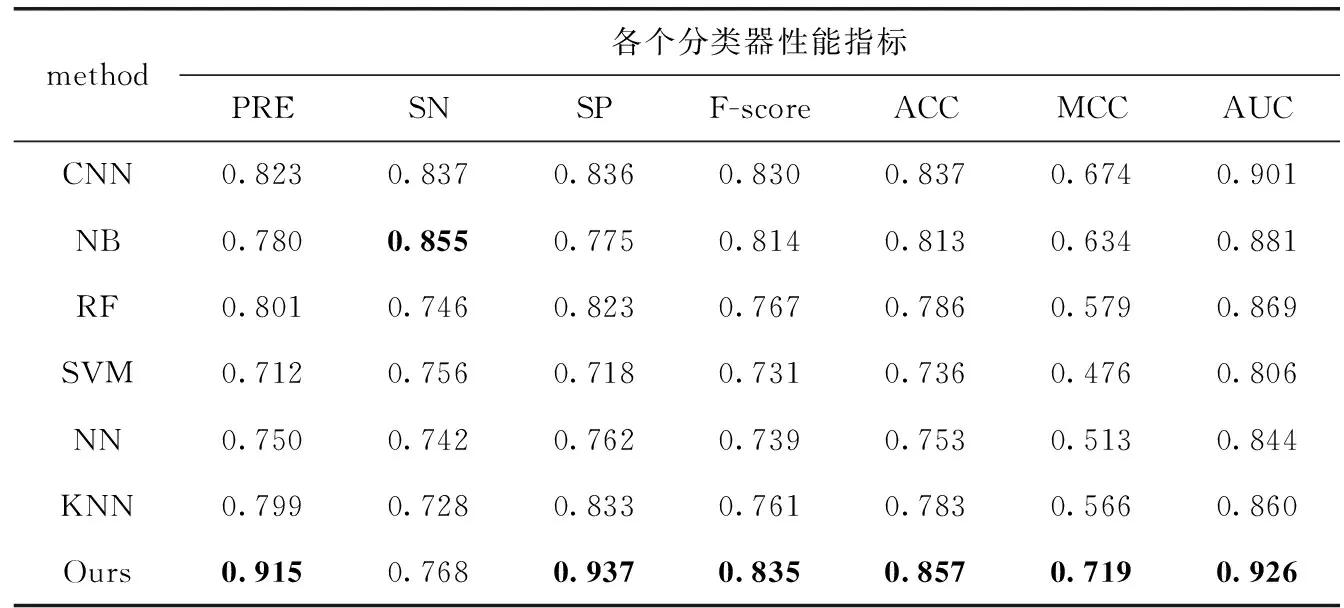
表3 革兰氏阴性细菌上的性能比较
2.6 在独立测试集上的预测结果
我们在一个独立的数据集上用我们的模型与其它4种模型(DeepT3-1、EffectiveT3、BPBAac和BEAN2)分别进行了性能比较。结果表明,与其它4种方法相比,我们提出的模型具有更高的敏感度(SN)、特异性(SP)、F值(F-score)、准确率(ACC)、马修相关系数(MCC),它们分别达到了0.943、0.965、0.930、0.951、0.901。它的准确率(ACC)为0.951,比DeepT3-1、EffectiveT3、BPBAac和BEAN2分别提高了2.5%、18.4%、8.0%、8.9%。其中我们模型的精确率(PRE)略低于BPBAac方法,BPBAac的精确率(PRE)达到了0.944,略高于我们的方法。与其它4种模型的性能比较具体见表4。
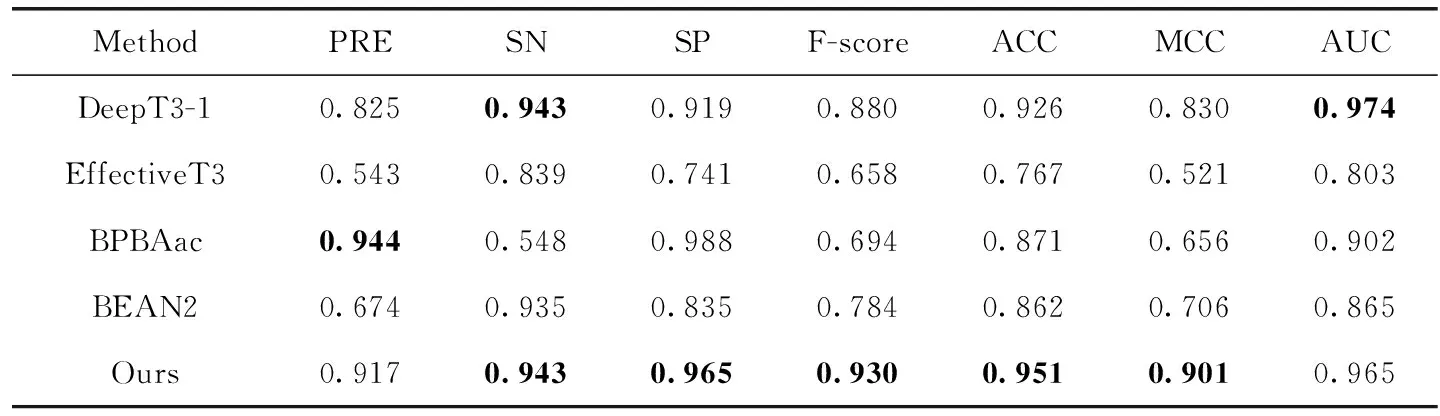
表4 不同模型比较结果
3 结束语
本文中,我们使用深度学习方法从一级序列中预测丁香型假单胞菌III型分泌效应蛋白。此方法跳过从序列对象中提取特征的步骤,能在训练过程中重新精确学习先导序列的已知基序。此外,该模型能在不同数据集上更准确地识别III型分泌效应蛋白。这项关于预测III型分泌效应蛋白的深度学习模型的全面研究会为未来的蛋白质组学研究提供一个有竞争力的工具。所提出的深度学习方法将有许多其它潜在的应用,如蛋白质远程同源性检测、miRNA预测等。
猜你喜欢
现代电力(2022年2期)2022-05-23
北京航空航天大学学报(2021年9期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
兽医导刊(2020年13期)2020-12-31
电子制作(2019年13期)2020-01-14
电子制作(2019年19期)2019-11-23
电子制作(2019年11期)2019-07-04
电子制作(2019年24期)2019-02-23
北京航空航天大学学报(2018年1期)2018-04-20
中成药(2017年3期)2017-05-17
