
HP0953 - hypothetical virulence factor overexpresion and localization during Helicobacter pylori infection of gastric epithelium
2022-08-12 08:10NancyArteagaResendizGerardoRodeaRosaMariaRibasAparicioAlmaOlivaresCervantesJuanArturoCastelVegaJosdeJesusOlivaresTrejoSandraMendozaElizaldeEdgarLopezVillegasChristianColinPamelaAguilarRodeaAlfonsoReyesLopezMarcelaSalazar
World Journal of Gastroenterology 2022年29期
Nancy K Arteaga-Resendiz,Gerardo E Rodea,Rosa Maria Ribas-Aparicio, Alma L Olivares-Cervantes,JuanArturo Castelén-Vega, José de Jesus Olivares-Trejo, Sandra Mendoza-Elizalde, Edgar O Lopez-Villegas,Christian Colin, Pamela Aguilar-Rodea,Alfonso Reyes-Lopez,Marcela Salazar Garcia,Norma Velazquez-Guadarrama
Abstract
Key Words: Hypothetical protein HP0953; Adherence; Helicobacter pylori; Glycocalyx; Ⅴirulence factor;Persistence
lNTRODUCTlON
The estimated prevalence ofHelicobacter pylori(H. pylori) infection is 70% in developing countries and 30%-40% in the United States and other industrialized countries[1]. However, not all people infected with this bacterium have symptoms.H. pylorihas been associated with gastroduodenal diseases and it is considered the major risk factor for the development of gastric cancer[2].H. pyloricolonizes the gastric mucosa, adhering to the mucous epithelial cells and to the mucous layer that covers the gastric epithelium. Adherence to gastric cells is the first step in establishing an infection and for successful colonization, the bacterium releases various effector proteins/toxins[3-5].
Upon colonization, the pathogenic mechanism ofH. pyloriis mediated by virulence factors such as flagella and the ability to form a biofilm (bothin vitroandin vivo)[6]. Biofilm formation enhances the ability of microorganisms to survive in a hostile environment, which would cause and exacerbate infections, rather than extend them, resulting in chronicity[7]. The glycocalyx participates in adhesion to inert surfaces, allowing biofilm formation but also participates in cell attachment; the major components of the glycocalyx are glycans and may contain digestive proteins or secreted virulence factors[8,9].H.pylorisecretes various virulence factors, including urease, cag pathogenicity island proteins,hemagglutinin, Lewis antigens (Lexand Ley), BabA CagA, and VacA[10-12]. All these virulence factors have been extensively investigated elsewhere.
Furthermore,H. pyloricontains hypothetical proteins, some of which are secreted particularly in the context of interactions with the host. The bacterium uses a set of secreted and translocated proteins to adapt itself to the mucosal environment[13].H. pyloripossesses a relatively small genome of < 1.6 Mb[14,15] compared with other gram-negative prokaryotes, such asEscherichia coli, whose genome is > 4.5 Mb[16]. Despite its small size, an ineligible fraction ofH. pyloriproteins, possibly 30%-40%, are annotated as “hypothetical proteins”. Using bioinformatics tools, Naqviet al[17] and Zanotti and Cendron[18] respectively analyzed 340 hypothetical proteins ofH. pylori26695. The function of some of these proteins can be hypothesized based on a weak homology with proteins of other bacteria, whereas most proteins do not exhibit similarities with others, and their function cannot be predicted[17,18].
HP0953 is a hypothetical and uncharacterized protein ofH. pylorisecretome; it lacks conserved domains but, interestingly, is present in allH. pyloristrains whose genomes have been sequenced, as well as inH. acinonychisthat colonizes the gastric mucosa of large felines. The closely relatedH. hepaticusandCampylobacter jejunilack HP0953 homologs, suggesting that HP0953 is unique to the gastricHelicobacterspecies[11]. Moreover, we previously observed that this protein is overexpressed during adhesion to inert surfaces under stress conditions. Most of the possible effects of secreted proteins on the host have yet to be discovered. In addition, secreted factors represent eligible proteins for identifying promising targets for the development of new antimicrobial drugs[17,18].
In recent years, there has been an increasing importance onH. pylori; on the one hand when it is present, it is associated with gastric disease, and on the other hand, it is associated with gastroduodenal reflux, food allergy, and asthma when it is absent[19]. This dual role in the human microbiota emphasizes the need for further characterization of the hypothetical proteins already described. In this study, the location of the protein is determined for the first time, inside and outside the bacterium, and we predict some biochemical characteristics, throughin silicoanalysis that contribute toward understanding the importance of this hypothetical protein to the microorganism in the establishment of infection, its role in such infection, or colonization of the host.
MATERlALS AND METHODS
In silico analysis of HP0953 protein
We obtained the amino acid sequence of HP0953 (accession number NP_207745) from the National Center for Biotechnology Information database[20]. Then, we used the Pfam database to analyze the HP0953 sequence and search for conserved domains or homologous regions present in other proteins[21].
Signal peptide prediction
The amino acid sequence of HP0953 was analyzed using the SignalP-4.1 tool provided by the ExPASy platform to predict the presence of a signal peptide[22].
Prediction of transmembrane helices and glycosylation sites
The amino acid sequence previously obtained was analyzed to predict transmembrane helices using the TM pred tool provided by the ExPASy platform[23,24]. The glycosylation sites were investigated using the GLYCOPP platform[25].
Three-dimensional modeling of HP0953 protein
To explore the remote homologs that could be used as a template for modeling, we used the HHpred tool that uses the HMM comparison[26]. Different programs such as I-TASSER[27], RaptorX[28], and HHalign-Kbest[29] were used to perform three-dimensional (3D) modeling.
Strain, cell culture, and extraction of genomic DNA from H. pylori
H. pyloristrain 26695 was cultured on Casman agar (Dibico; Mexico) supplemented with 5% sheep erythrocytes and incubated in an atmosphere of 50 mL/L CO2and 37 °C for 48 h. The culture was harvested for infected human against gastric (AGS) cell-line assay and genomic DNA extraction.Genomic DNA extraction was performed according to the protocol established for the Wizard kit(Promega; United States) by its manufacturer.
Amplification and purification of HP0953
HP0953was amplified by polymerase chain reaction (PCR) usingH. pylorigenomic DNA as the template. To amplify the complete gene, 50 μL of reaction mixture was prepared as follows: 1.25 U polymerase GoTaq®, 0.2 mmol/L dATP, 0.2 mmol/L dGTP, 0.2 mmol/L dCTP, 0.2 mmol/L dTTP, 1.5 mmol/L MgCl2(Promega), HP0953F oligonucleotide (25 pmol) (5’-GGGGGATCCATGGTTTTAATCGCTCTTTTAGGGGTG-3’), HP0953R oligonucleotide (25 pmol) (5’-GGGGTCGACCCTTAACGCACAAACGCTACC-3’), andH. pylori26695 DNA (250 ng). For amplification of the gene without the signal peptide, the same PCR was performed substituting the oligonucleotide HP0953F by HP095WSPF (5’-GGGGGATCCGTCTCTGATTTAAAGGGCATG-3’). The amplification cycle consisted of 94 °C for 1 min, followed by 30 cycles at 94 °C for 30 s, 68.4 °C for 30 s, 72 °C for 1 min, and a final step at 72 °C for 7 min. Both PCR products (555 and 474 bp) were purified using the protocol established by Promega for using the Wizard®SV gel and the PCR Clean-Up System (Promega).
Cloning of the complete and without-signal peptide hp0953
CompleteHP0953and its version without the signal peptide were inserted into the vector pJET1.2(Thermo Scientific; Lithuania) according to the manufacturer’s instructions to obtain the recombinant pJET-HP0953 and pJET-HP0953WSP plasmids, respectively. Chemically competent DH5α cells ofEscherichia coli(E. coli) were transformed with each plasmid by thermal shock[30,31]. The recombinant colonies were selected in LB agar (Sigma-Aldrich; MO) containing 100 μg/mL ampicillin. The plasmids were extracted using the illustraTMplasmidPrep Mini Spin Kit (GE Healthcare; United Kingdom).
The pJET-HP0953 and pJET-HP0953WSP plasmids were digested with the fast restriction enzymes BamHI and Sal I according to the manufacturer’s instructions (Thermo Scientific) to free both versions ofHP0953. Restriction products were purified using the protocol established by Promega for the Wizard®SV gel and the PCR Clean-Up System. DNA fragments were ligated into the expression vectors pET28a(+) (Novagen, United States) for the completeHP0953and pGEX-6p-2 (GE Healthcare) for the version without the signal peptide. The vectors were digested with the same enzymes and treated with alkaline phosphatase (Thermo Scientific). The Rapid DNA Ligation Kit (Thermo Scientific) was used to obtain the recombinant pET-HP0953 and pGEX-HP0953WSP plasmids. Chemically competent DH5α cells ofE. coliwere transformed with each plasmid by thermal shock. The recombinant colonies were selected in LB agar containing 50 μg/mL kanamycin for pET-HP0953 or 100 μg/mL ampicillin for pGEX-HP0953WSP. The plasmids were extracted using the illustraTMplasmidPrep Mini Spin Kit (GE Healthcare) and analyzed by PCR. DNA inserts in the recombinant plasmids were characterized by sequencing; for this purpose, PCRs were performed as described earlier using the following primers:T7F (5’-TAATACGACTCACTATAGGG-3’) and T7R (5’-GCTAGTTATTGCTCAGCG-3’) for pETHP0953 and HP095WSPF (5’-GGGGGATCCGTCTCTGATTTAAAGGGCATG-3’) and HP0953R (5’-GGGGTCGACCCTTAACGCACAAACGCTACC-3’) for pGEX-HP0953WSP. The annealing temperature was 68.4 °C for pET-HP0953 and 60 °C for pGEX-HP0953WSP. The PCR products were purified as described earlier and sequenced by Synthesis and Sequencing Unit, Institute of Biotechnology,Universidad Nacional Autónoma de México. Chemically competent BL21 pLysS cells ofE. coliwere transformed with each recombinant plasmid and the empty plasmids by thermal shock[31]. The recombinant colonies were selected in LB agar containing the corresponding antibiotic.
Expression and purification of HP0953 protein
The HP0953 protein was expressed according to the manufacturer’s instructions for pET28a(+) and pGEX-6p-2 vectors using 1 mmol/L IPTG (Sigma-Aldrich) and incubating theE. colipET28a(+)/pETHP0953-transformed cells for 16 h at 30 °C to obtain the recombinant protein His-HP0953 and theE. colipGEX-6p-2/pGEX-HP0953WSP-transformed cells for 3 h at 37 °C to obtain the recombinant protein GST-HP0953WSP. The inclusion bodies were extracted as described in the inclusion body preparation method without solubilizing the inclusion bodies in a denaturing agent[32]. The cell pellet was resuspended in 1 mL of 4 × sample buffer. Aliquots were collected throughout the process and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot assays.The inclusion bodies were electrophoresed on SDS-PAGE, and the recombinant protein band was purified by electroelution. The glutathione-S-transferase (GST)-HP0953WSP recombinant protein was used to produce polyclonal antibodies, as described later, and the His-HP0953 protein was used to purify antibodies.
Production of polyclonal anti-GST-HP0953WSP
Anti-GST-HP0953WSP antibodies were obtained by immunization of a New Zealand rabbit (2 mo age)according to the Howard and Bethell method[33] (approved by the Bioethics Committee of the Hospital Infantil de México Federico Gómez).
Purification of polyclonal anti-HP0953
Purification of polyclonal antibody was performed by immunoadsorption. The His-HP0953 protein was subjected to SDS-PAGE on preparative gels. The bands were transferred onto a polyvinylidene fluoride(PVDF) membrane (Millipore; United States), which was blocked with 0.5% bovine serum albumin(BSA) and washed with phosphate buffer solution (PBS)-Tween. Serum was diluted 1:100 in PBS-Tween,incubated with the membrane for 2 h under stirring at room temperature, and washed with PBS-Tween;then, the membrane was incubated with 0.02 M glycine-HCl (pH 2.2) for 10 min at room temperature.Finally, 1 M Tris base (pH 9.1) was added. The antibody was recovered and stored at -20 °C.Nonpurified antisera and purified antibodies were tested against a total extract ofH. pylori, and the two purified recombinant proteins were evaluated by western blotting using the same PVDF membrane,which was stripped between each test[34].
Construction of plasmid pBlue-5CM3 for site-directed mutagenesis of the HP0953 gene in H. pylori
The chloramphenicol resistance gene was obtained from the pLysS plasmid of theE. coliBL21 (DE3)pLysS strain by PCR, for which 50 μL of reaction mix was prepared, consisting of 1 × HF Fusion buffer(Thermo Scientific); 0.2 mmol/L each of dATP, dGTP, dCTP, and dTTP (Promega); 6 pmol each of primer CMF (5’-GGG AGATCTTTACGCCCCGCCCTG-3’) and CMR (5’-GGGCATATGATGGAGAAAAAAATCACTGG-3’); plasmid pLysS (250 ng); and Phusion DNA Polymerase (1 U) (Thermo Scientific). The CMF and CMR primers contain restriction sites forBglII andNdeI, respectively. The amplification program consisted of initial denaturation at 98 °C for 30 s; 35 cycles of denaturation at 98°C for 10 s, primer annealing at 65 °C for 30 s, and extension at 72 °C for 1 min; and a final extension at 72 °C for 7 min.
The 5’ and 3’ fragments of theHP0953gene were obtained from the plasmid pJET-HP0953,constructed above by PCR. To amplify the 5’ fragment, 50 μL of the reaction mixture was prepared,consisting of GoTaq DNA polymerase (Promega) (1.25 U); 0.2 mmol/L each of dATP, dGTP, dCTP, and dTTP; 1.5 mmol/L MgCl2(Promega); primer HP0953F (25 pmol) (5’-GGGGGATCCATGGTTTTAATCGCTCTTTTAGGGGTG-3’); primer HP0953R (5’-GGGAGATCTCATGGAATTGCTCCATGAAGCG-3’); and plasmid DNA from pJET-HP0953 (250 ng). The amplification program consisted of initial denaturation at 94 °C for 1 min; 35 cycles of denaturation at 94 °C for 30 s, primer annealing at 68.4 °C for 30 s, and extension at 72 °C for 30 s; and a final extension at 72 °C for 7 min. To amplify the 3’fragment, PCR was performed under the same conditions but using the primers HP0953F (5’-GGGCATATGAGTCCGAGCAGACGACTACG-3’) and HP0953R (5’-GGGGTCGACCTACCTTAACGCACAAACGCTACC-3’). Amplicons of 304 and 248 bp were obtained, respectively.
We subjected 5 μL of each amplification product to 1.5% agarose gel electrophoresis at 85 volts for 75 min. Then, we stained the gel with ethidium bromide (Sigma-Aldrich) and observed the bands under ultraviolet light. Upon confirmation of the amplified product, the remainder was purified using a Wizard SV Gel and PCR Clean-up System, as per the manufacturer’s protocol. Each fragment was inserted into a pJET1.2 vector. The ligation reaction of each fragment in the pJET1.2 vector consisted of 1× reaction buffer, 0.15 pmol purified PCR product (5’ fragment, 3’ fragment, or chloramphenicol resistance gene; 0.15 pmol), pJET1.2/Blunt Cloning Vector (0.05 pmol), and T4 DNA ligase (5 U). The reaction mixture was incubated at 22 °C for 20 min.
After ligation, we used heat shock treatment to transform 50 μL of competentE. coliDH5α cells with each plasmid[31]. Then, they were centrifuged at 5000 × g for 1 min, the supernatant was decanted, and the pellet was resuspended in the residual medium. Subsequently, the bacterial suspension was seeded on LB agar plates containing ampicillin (100 μg/mL; Sigma-Aldrich) and incubated at 37 °C for 18-24 h to select the recombinant clones. Following incubation, we selected five colonies from the plate and resuspended them in 2 mL of LB broth (Sigma-Aldrich) containing ampicillin (100 μg/mL), and the plasmid was extracted using the alkaline lysis method[35]. We quantified the plasmid DNA using an Epoch microplate spectrophotometer (Biotek Instruments, Inc.; United States) and stored it at -20 °C.
We performed three PCR assays to verify the sequence of the cloned fragments. Each PCR reaction was performed in a final volume of 20 μL, consisting of 1 × HF Fusion buffer (Thermo Scientific); 0.2 mmol/L each of dATP, dGTP, dCTP, and dTTP (Thermo Scientific); primer pJET1.2F (0.2 μM) (5’-CGACTCACTATAGGGAGAGCGGC-3’) (Thermo Scientific); primer pJET1.2R (0.2 μM) (5’-AAGAACATCGATTTTCCATGGCAG-3’) (Thermo Scientific); plasmid DNA (100 ng/μL) (pJET-CM,pJET-5’ or pJET-3’); and Phusion DNA polymerase (Thermo Scientific) (0.4 U). The amplification program consisted of initial denaturation at 98 °C for 30 s; 35 cycles of 98 °C for 10 s, 68 °C for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 7 min. The PCR products were sequenced by Macrogen(Seoul, Korea).
The 5’ fragments, 3’ fragments, and the chloramphenicol resistance gene were obtained from the plasmids previously constructed by enzymatic digestion.BamHI andBglII were used for the 5’fragment,NdeI andSalI for the 3’ fragment, andBglII andNdeI for the chloramphenicol resistance gene,according to the above-described digestion protocols. The fragments were inserted into the pBluescript II KS+ vector (Agilent Technologies, Inc.; United States), which allows for the synthesis of ssDNA and has an ampicillin resistance gene to facilitate selection following the above-described protocols.E. coliXL1-blue MRF’ cells were transformed with the constructed vector (pBlue-5CM3) using heat shock treatment and selected following culture on LB agar plates (Sigma-Aldrich) with 100 μg/mL of ampicillin[31]. The plasmid was extracted using alkaline lysis[35], and fragment insertion was verified by automated sequencing.H. pyloriwas transformed with the plasmid using electroporation[36]. The mutantH. pyloristrain was cultured on Casman agar supplemented with 5% sheep erythrocytes and incubated in an atmosphere of 50 mL/L CO2at 37 °C for 48 h.
Identification of HP0953 in H. pylori
The purified antibody was used for identifying the protein HP0953 inH. pylori26695 by western blotting. A total extract obtained fromH. pyloriadhesion test (see below) was used, and soluble and insoluble fractions were separated according to the inclusion body preparation method[32]. These fractions were subjected to SDS-PAGE (50 mg of protein) and transferred onto a PVDF membrane,where western blotting was performed using the purified antibody diluted 1:2 in PBS-Tween and incubated overnight under stirring at 4 °C and the secondary antibody diluted 1:10000 [anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP)-conjugated antibody] (Santa Cruz Biotechnology; United States)[34]. Washing was done with TBST (2 mmol/L Tris, 15 mmol/L NaCl, 0.1%Tween 20, pH 7.6). Finally, the membrane was visualized using Immobilon Western Chemiluminescent HRP substrate (Millipore).
Adhesion test of H. pylori
A suspension (6 × 108) ofH. pylori26695 culture in Brucella broth (Dibico) supplemented with 1% yeast extract was prepared. Next, 50 mL of the suspension was placed in cell culture bottles and incubated at 37 °C in an atmosphere of 50 mL/L CO2for 0, 1, 3, 12, and 24 h; these time points are the first hours of biofilm formation where adherence occurs. After the incubation period, the supernatant was removed,and the bottle walls were scraped to obtain adherent cells, which were resuspended in 1 mL of sterile 1× PBS.
Thawing and seeding of AGS cells
First, 1 mL of AGS cell suspension was placed in a 15 mL conical tube. Subsequently, 2 mL of DMEMF12 (Gibco, Life Technologies; United States) medium was added for washing by centrifugation at 1500 rpm for 5 min. A second washing step was performed, and the cells were resuspended again in DMEMF12 medium. Next, 1 mL of the cell suspension was taken to seed into cell culture bottles. The bottles were incubated in a CO2chamber for 24 h at 37 °C.
AGS cell infection assay by H. pylori
The gene expression assay during biofilm formation was performed in duplicate in ten cell culture bottles of 225 mL each. A suspension ofH. pylori(strain 26695, previously cultured for 24 h in a plate with Casman medium) was prepared in DMEM-F12 culture medium. The cells were washed by centrifugation at 3000 rpm for 15 min. After washing, the cell pellet was used to prepare a bacterial suspension in 300 mL of DMEM-F12 culture medium inside a flask until the growth level reached similar to that in tube number 2 on the McFarland scale. To each bottle, 30 mL of the bacterial suspension similar to McFarland tube 2 suspension was added, and then 20 mL of DMEM-F12 medium without the bacteria was added to complete 50 mL. One bottle was not infected with the bacterial strain and was used as the control for the transmission electron microscopy (TEM) assay. The bottles were incubated at 3, 12, 24, and 48 h post-infection. Time 0 was taken as the culture of the bacterial strain with no contact with AGS cells for the expression assay. After each time point of incubation, the supernatant was discarded, and 1 mL of sterile 1 × PBS was added for washing the cells. Subsequently, the wall of the bottle was scraped with a gendarme to detach the cells and remove the adhered biofilm. Each sample was processed for RNA extraction assay. An aliquot was taken for use in TEM, and the remainder was centrifuged at 7000 rpm for 15 min. The test was conducted in triplicate.
Immunogold technique for TEM
First, 500 μL of the samples obtained from the adhesion test was centrifuged at 2500 rpm for 10 min and fixed with 4% paraformaldehyde (MP Biomedicals; United States) in 1 × PBS (pH 7.4) for 1 h at 4 °C.Then, three washes were performed with 1 × PBS (pH 7.4), and the sample was dehydrated by adding 50% ethanol (Merck; IRL) for 10 min, followed by two washes with 70% ethanol for 15 min at room temperature. The inclusion was done using the LR-White method as described by Vázquez and Echeverría[37], and ultrathin sections of 60-90 nm were obtained. The sections were placed on nickel grids, blocked with 5% BSA for 20 min, and incubated with the purified polyclonal antibody overnight at 4 °C. The sample was then washed with TBST and incubated with the secondary antibody (goat antirabbit IgG coupled to 10-nm colloidal gold particles, 1:5, Sigma-Aldrich) diluted in deionized water for 1 h at room temperature. The grids were rinsed with TBST. Finally, they were observed under a transmission electron microscope (JEM 1010 JEOL Ltd. Japan) equipped with a TEM Imaging Systems AMT (Woburn; United States).
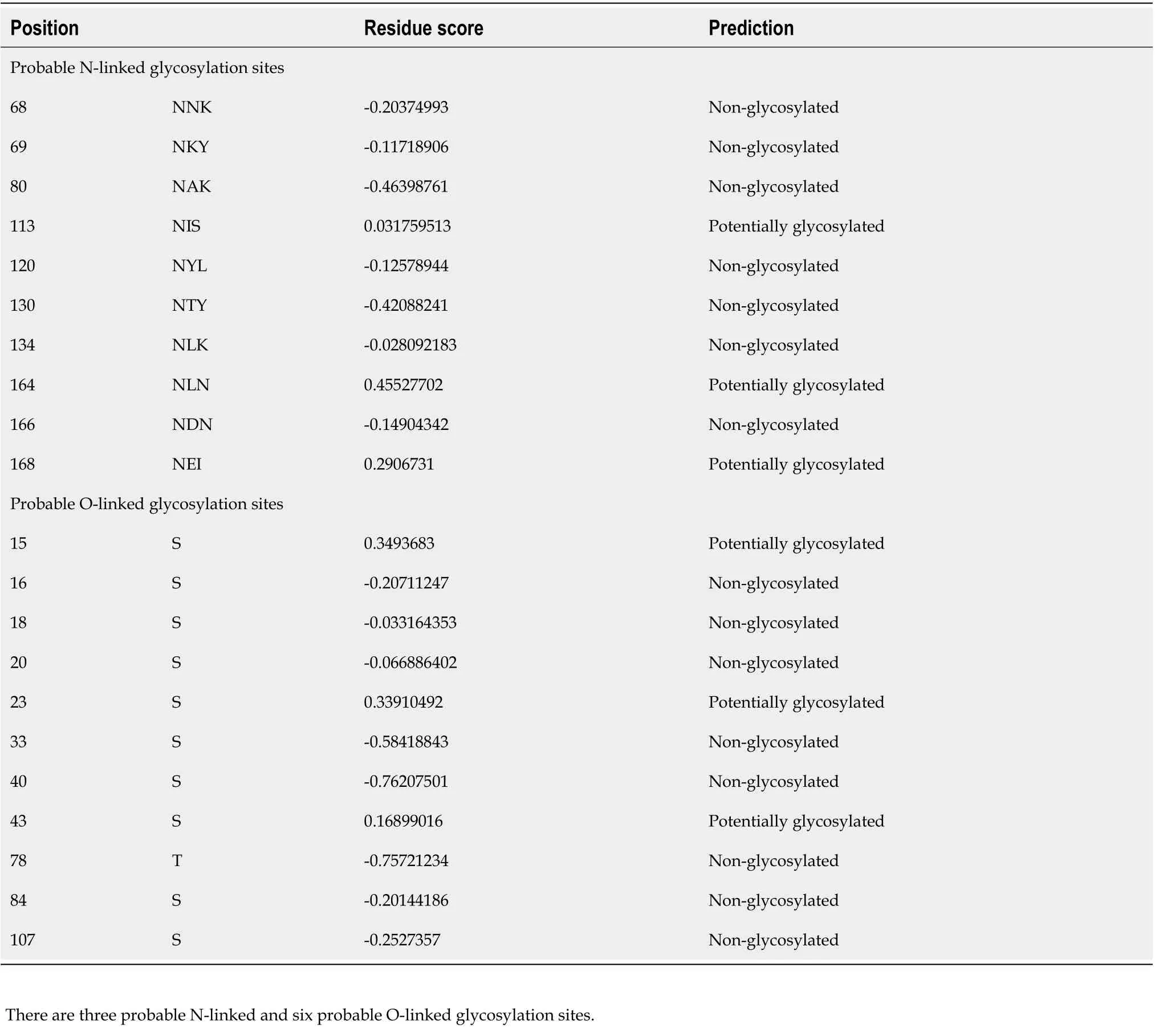
Table 1 Prediction of glycosylation sites
Quantitative SYBR-Green real time-PCR analysis for hp0953 expression during H. pylori infection of AGS cells
Expression analysis was performed by real time-PCR (RT-qPCR). The glutamate racemase (glmM)gene was used as an internal control for the bacterial cell, and theβ-globingene was used as an internal control for human AGS cells.HP0953expression was evaluated at different time points (0, 3, 12, 24, and 48 h). Time 0 was taken as the culture of the bacterial strain with no contact with AGS cells.
Gene-specific primers were developed forHP0953andglmMusing the Primer3 software, v.0.4.0[38,39]. The sequences of all the primers were as follows:HP0953(5’-CATATGCCGAGCAGACGACTACG-3’; 5’-GTCGACCTACCTTAACGCACAAACGCTACC-3’),glmM(5’-ACCGACGCTCTCACCCACTT-3’;5’-AGCGCGAGCCACAACCCTTT-3’). Total RNA was isolated from the AGS cell cultures with and withoutH. pyloriat a multiplicity of infection of 1:100. RNA was extracted using TRIzol®Reagent(Ambion RNA, Life Technologies; United States), followed by phenol:chloroform extraction. The quality and amount of the resulting RNA were evaluated using a Nano-Drop spectrophotometer (The Epoch™Multi-Volume Spectrophotometer System; United States) at 260 and 280 nm, respectively.
Reverse transcription reaction and SYBR-Green PCR were performed as described below. To initiate the first-strand cDNA synthesis, 1 mg of total RNA was treated with 500 μL of DNAse I (10 U/mL;Invitrogen; United States). Then, it was incubated for 30 min at 37 °C and 5 min at 75 °C with 4 μL of 5 ×RT buffer, 2 μL of 25 mmol/L MgCl2, 2 μL of 10 mmol/L dNTP mix, 2 μL of 50 mmol/L random hexamer primers, 1 μL of RNAse inhibitor (20 U/μL), and sufficient H2O for a final volume of 20 μL.The mixture was then incubated for 10 min at 25 °C and 30 min at 48 °C with 2 μL of MultiScribe reverse transcriptase (50 U/mL; Applied Biosystems; United States). The reaction was terminated by heating the sample at 95 °C for 10 min. Aliquots of 200 ng of cDNA were used in the SYBR-Green PCR analysis according to the manufacturer’s protocol. RT-PCR amplification was performed in triplicate under the following conditions: 2 min at 50 °C (for UNG activation) and 10 min at 95 °C (for polymerase activation), followed by 40 cycles of denaturation and alignment/extension at 95 °C for 15 s and 60 °C for 1 min, respectively, using the Stratagene Mx3005p qPCR System and MxPro-Mx3005p software(Santa Clara; United States). Relative quantification of HP0953 RNA expression was performed using the comparative CT method. The ΔCt (difference in CT values) between HP0953 and theglmMinternal control and the ΔΔCt were calculated to normalize the differences in cDNA concentrations for each reaction. The RNA expression level was calculated using the equation 2-(ΔΔCt)[40]. The Kruskal-Wallis equality-of-populations rank test was used to compare the expression of the gene at all study time points. Differences were considered to be significant when thePvalue was < 0.05.
RESULTS
In silico analysis of HP0953 protein features
In silicoanalyses allow us to foresee probable protein maturation. The analysis conducted using the SignalP 4.1 tool revealed that HP0953 protein loses a signal peptide after excision between amino acids 21 and 22 which could lead to extracellular release (Figure 1). This region is also believed to present a transmembrane helix (Figure 2). The protein modifications provided us an insight into their probable function. Tables 1 and 2 show the predicted modification sites for HP0953. There are three predicted Nlinked glycosylation sites in amino acid positions 113, 164, and 168 and six predicted O-linked glycosylation sites in positions 15, 23, 43, 111, 115, and 183. Moreover, there are four predicted myristoylation sites in positions 12-17, 29-34, 73-78, and 182-187 and one predicted prenylation site in the position 185-188.
To predict the structural model, we used different programs such as I-TASSER, RaptorX, and HHalign-Kbest, the latter being the one that generated a better quality model (Figures 3 and 4). The HHpred program disclosed a similarity with the Tip-alpha protein ofH. pylori; thus, it was used as a template for modeling. Additionally, no conserved domains were found or matches with other proteins.
Site-directed mutagenesis of HP0953 gene
Plasmid pBlue-5CM3 was constructed forHP0953site-directed mutagenesis (Supplementary Figure 1).Sequencing the Dhp0953::CmBlue-5CM3 construction multiple cloning site corresponded to insertedHP0953andCmcassette. However, we were unable to propagate theH. pylori-transformed strain.
HP0953 is detected in the insoluble fraction of H. pylori protein extract
ForH. pyloriHP0953 protein identification, insoluble and soluble fractions were obtained and resolved by SDS-PAGE assay. HP0953 protein was not detected in the soluble fraction, and its total concentration was very low in the insoluble fraction (data not shown). Interestingly, HP0953 was expressed constitutively at all the tested time points (Figure 5).
HP0953 is located in the cytoplasm and the peripheral zone of H. pylori
Protein location is indicative of its function. HP0953 was identified in the insoluble fraction ofH. pylorilysates, which probably implies that the function of HP0953 is elicited in the vicinity of the outer membrane. To examine this hypothesis, we performed the immunogold technique on bacterial sections that were analyzed by TEM. The micrographs showed that HP0953 protein is located in the cytoplasm and accumulated in some peripheral areas of the bacterial body (Figure 6A), as well as in bacteriabacteria contact areas (Figures 6B, 6C and 6D). Furthermore, there was a large accumulation of the protein in the bacterial debris (Figure 6E). Interestingly, the presence of this protein was also observed in the area of the glycocalyx that coversH. pylori(Figure 7), and accumulated where said coating spreads, producing a type of net, perhaps indicating the formation of a biofilm. Interestingly, a large amount of HP0953 protein was secreted during the infection of gastric cells because it was found in theH. pylori-AGS cell contact area (Figure 8).
Higher expression of HP0953 is required in the early stages of H. pylori-AGS cell interaction
RT-qPCR was performed to elucidate the behavior ofHP0953during the contact ofH. pyloriwith AGS cells. Figure 9 shows a comparison of the expression ratios of the treated and untreated cultures.HP0953expression was observed at all the tested time points; however, higher expression values were observed after 12 h of infection.glmMwas the prokaryotic constitutive gene that was used as an internal control(Figure 9).
DlSCUSSlON
The high prevalence ofH. pyloriinfection and its significant persistence in the gastrointestinal tract in the population with limited economic resources, as well as the diversity of related pathologies, theenvironment, and the genetics of the host, suggest that the pathogenicity mechanisms used by this microorganism are numerous. The different bacterial strains express variations in their virulence factors,including those associated with the colonization process (e.g.,urease, flagella, a chemotaxis system, and adhesins), immune response evaders (e.g.,lipopolysaccharide and flagella; CagA and the type IV secretion system, VacA; gamma-glutamyl transpeptidase, and catalase superoxide dismutase), and disease inducers (e.g.,VacA, BabA, IceA, HtrA, CagA, and the type IV secretion system, DupA, and OipA)[41]. Furthermore, theH. pyloriproteome consists of 340 proteins that are described as hypothetical, implying that their structure or function is unknown, thus indicating that some proteins may be related to pathogenicity mechanisms or are important to adapt to the surrounding environment.The HP0953 protein exhibited its maximum expression at the first 12 h of infection in a culture of gastric epithelium cells; this finding along with its location near the infected cells and the predicted prenylation and myristoylation sites suggest that it is an important protein during the bacterium’s colonization phase of the gastric epithelium.

Table 2 Prediction of posttranslational modification sites

Figure 1 Prediction of a signal peptide for HP0953 protein. The green line that represents the S-score indicates a high probability that the first 20 amino acids conform a signal peptide that can be removed by a cut between amino acids 20 and 21, as marked by the tallest red line indicating the C-score.

Figure 2 Prediction of transmembrane helices. Values > 0 indicate the probable presence of transmembrane helices, as observed in the region constituted by the first 25 amino acids. The HP0953 protein could be anchored to the membrane to be subsequently secreted.
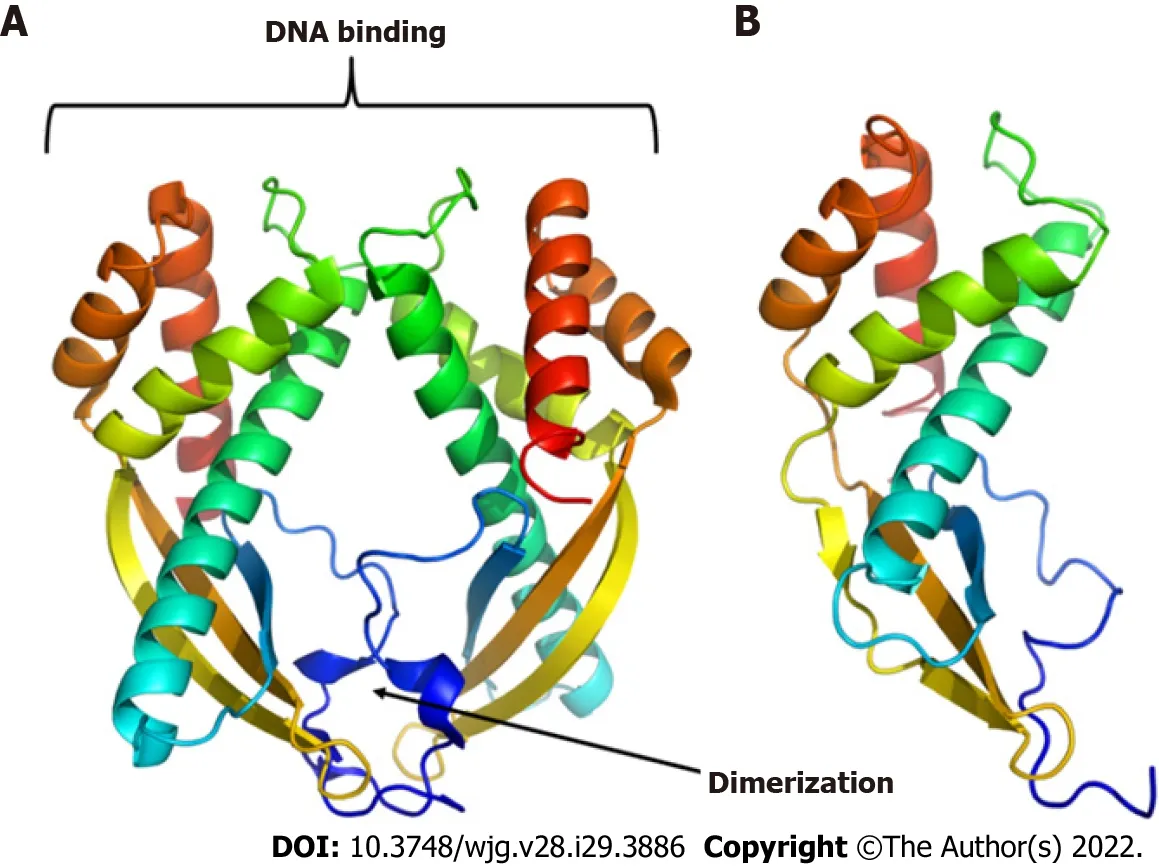
Figure 3 Three-dimensional models of proteins. A: Tip-alpha protein from Helicobacter pylori (H. pylori); B: HP0953 protein from H. pylori. Tip-alpha acquires a dimer arrangement that could also be present in the HP0953 protein due to its structural similarity in the position of the alpha helices and beta sheets.
Although various studies, such as those conducted by Naqviet al[17] in 2016 and Parket al[42] in 2012, have performedin silicoanalyses to determine the probable structures and functions of the hypothetical proteins ofH. pylori, none of them evaluated the probable function of HP0953[17,42].Furthermore, the bioinformatics analysis performed in this study highlighted the presence of a signal peptide that may anchor at the transmembrane region and then allow the release of the protein to the external environment after its excision. The analyses also revealed some probable glycosylation sites,which suggests that HP0953 is a protein from the glycocalyx. The glycocalyx is defined as any glycoprotein-associated polysaccharide that contains bacterial surface structures and is distant from the outer membrane or cell wall surface[43]. Bacterial glycocalyx generally has protection functions that allow bacterial survival and persistence in the natural environment and cell attachment[44].
Different modification patterns are found within the HP0953 sequence, such as the myristoylation pattern. This modification is essential for the removal of membrane proteins[45]; the existence of these putative sites provides an idea about its pathogenicity. Importantly, these bacteria lack the enzyme Nmyristoyl transferase (NMT) required for producing this modification; hence, the proteins that are myristoylated are processed by the NMT of their eukaryotic hosts[46]. NMT is a suitable therapeutic target in opportunistic infections in humans; in fact, it has been associated with carcinogenesis, in particular colon cancer[47]. Myristoylation has been suggested to occur as part of the secretory machinery[48,49]. Hypothetical patterns of prenylation were also detected, which is a class of modification involving the addition of isoprenoids or geranylgeranyl, and it is involved in membrane interactions due to the hydrophobicity of these lipids[50]. This modification is often a step to target the affected proteins to specific membranes[51]. As this modification was predicted in HP0953 by the ProtComB tool, it could be relevant for its function.
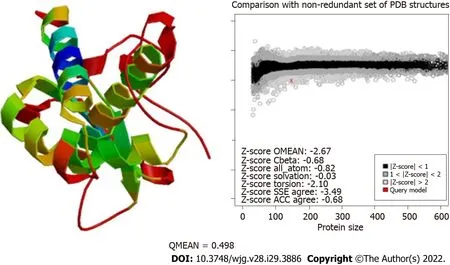
Figure 4 Evaluation of the modeling quality of HP0953. The QMEAN for this protein was 0.498, which indicates that the obtained model is acceptable.PDB: Protein Data Bank; SSE: Secondary Structure Element; ACC: Solvent Accessibility.

Figure 5 Location of the hypothetical protein HP0953 in the insoluble fraction of Helicobacter pylori 26695. A: Sulfate-polyacrylamide gel electrophoresis 16% gel stained with Coomassie blue; B: Western blot with an arrow showing the band corresponding to the expected molecular weight. Lanes: (1)Molecular weight marker and (2-5) insoluble fractions of Helicobacter pylori 26695 from samples collected at 0, 1, 3, and 24 h of incubation during biofilm formation.The arrow indicates the presence of the native HP0953 protein. H. pylori: Helicobacter pylori.
Extracellular proteins can play a vital role in bacterial pathogenesis[11], which correlates with our findings of the similarity between the HP0953 protein and the Tip-alpha protein, which is described as the only virulence factor secreted by allH. pyloristrains[52]. Moreover, its overexpression has been related to the development of gastric cancer. Tip-alpha is an exotoxin that penetrates cells and induces the production of tumor necrosis factor alpha[53]. HP053 protein could have a similar function because probable posttranslational modification sites related to bacterial pathogenesis were also found. It has been reported that some bacteria secrete effectors through the type III or type IV secretion systems,mimicking the functions of eukaryotic proteins to evade the defense systems. Subsequently, they undergo a posttranslational modification mediated by the same host enzymes to become biologically active[54], stimulating or inhibiting the immune response, interfering signaling pathways, affecting Tcell activation, or whatever their function may be[55]. Among these posttranslational modifications are myristoylation and prenylation[56,57], which were also detected in thein silicoanalysis conducted in this study. These modifications added lipid residues that allow the protein to anchor to the cell membrane to exert some unknown function.
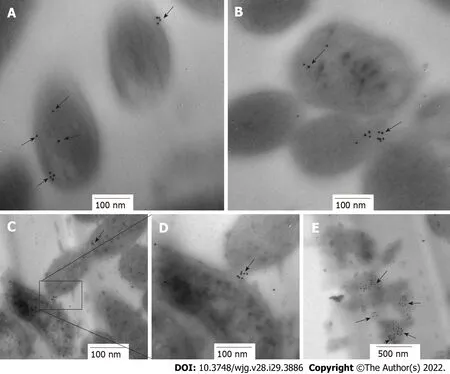
Figure 6 lmmunolocation of HP0953 protein in Helicobacter pylori bacterial transmission electron microscopy sections. A and B: 100000 ×;C: 75000 ×; D: 150000 ×; E: 60000 ×. The black arrows show the location of HP0953 protein. Immunogold technique, primary antibody (polyclonal rabbit anti-HP0953), secondary antibody (goat anti-rabbit immunoglobulin G coupled to 10-nm colloidal gold particles). (JEM 1010 JEOL Ltd. Tokyo, Japan) microscope equipped with a TEM Imaging Systems AMT (Woburn, MA, United States).
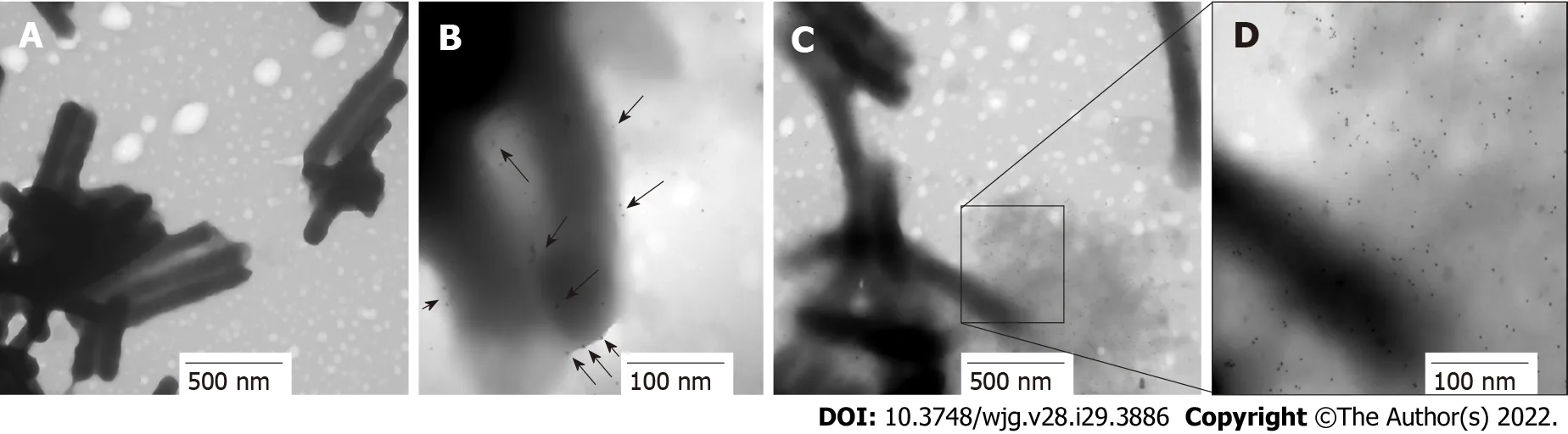
Figure 7 lmmunolocation of HP0953 protein in whole bacteria. A: Helicobacter pylori (H. pylori) incubated with the secondary antibody alone, 20000 ×; BD: Complete immunogold technique for H. pylori; B: 100000 ×; C: 30000 ×; D: 100000 ×. Black arrows indicate the location of HP0953 protein. Immunogold technique, primary antibody (polyclonal rabbit anti-HP0953), secondary antibody (goat anti-rabbit immunoglobulin G coupled to 10-nm colloidal gold particles).
HP0953 lacks homologous proteins outsideH. pylori, and this absence limits comparisons and the elucidation of its potential role. X-ray crystallography analysis can be performed to determine its 3D structure, which could then be used in comparisons with other proteins in the database. However, it was observed that HP0953 is an insoluble protein, which makes it difficult to purify in its native form.
The persistence ofH. pyloriin an environment where there are peristaltic movements and cell shedding is mediated by a variety of adhesins present on the bacterial surface[10,58], and the adhesion ofH. pylorican be achieved by a connection of the plasma membrane through filamentous materials.However, it is known that glycocalyx preferentially adheres to the surfaces of biomaterials and compromised tissues, forming biofilms[55]. Research on biofilm composition has shown that apericellular matrix, also known as the glycocalyx, develops to preserve and concentrate the lytic enzymes secreted by bacteria, which, in turn, increases their metabolic efficiency[8]. This suggests that the hypothetical protein HP0953 could be involved in lytic processes by being present in the glycocalyx and concentrated in the bacterial debris; however, functionality studies are still required to elucidate the role of this protein in the persistence ofH. pyloriin the human stomach. Smithet al[11] suggested that this protein could be involved in the survival of the bacterium in the gastric environment based on their finding that it is also present inH. acinonychis,which colonizes the gastric mucosa of felines.Nevertheless, other closely related ε-proteobacteria, such asH. hepaticusandC. jejuni, lack sequences that are homologous to HP0953, thus suggesting that this protein is unique to the gastricHelicobacterspecies[11], suggesting an important role of HP0953 inH. pylorigastric pathogenesis.

Figure 8 Location of HP0953 protein during against gastric cell infection by Helicobacter pylori. A: Against gastric (AGS) cell at 3 h of infection byHelicobacter pylori (H. pylori), incubated with the secondary antibody alone, 40000 ×; B: Complete immunogold technique for AGS cells at 3 h of infection by H. pylori,40000 ×; C: Complete immunogold technique for AGS cells at 3 h of infection by H. pylori, 50000 ×; D: Complete immunogold technique for AGS cells at 12 h of infection by H. pylori, 40000 ×. White arrows indicate the location of HP0953 protein.
In our previous study, we observed the overexpression of HP0953 protein during the first hours of adherence to inert surfaces and AGS cells[59], suggesting its involvement in the survival ofH. pyloriin the environment or in its pathogenic mechanisms. In the present study, we found that initial levels ofHP0953expression become diminished, perhaps as a result of the adaptation ofH. pylorito its environment. The levels of HP0953 rise after 3 h, indicating that this protein is required for the interaction ofH. pyloriwith AGS cells, and the maximum level ofHP0953expression occurred 12 h after the infection of AGS cells. Genetic regulation in pathogens allows for fluctuation in the expression of constitutive genes following initial contact with the target cells[60,61]. The immunogold assay demonstrated that HP0953 protein was located in the cytoplasm, accumulated in some peripheral areas of the bacterial body, and exhibited a greater expression when it is close to AGS cells. These data suggest that HP0953 protein is synthesized in the cytoplasm and subsequently exported to the outer membrane, where it is involved in adhesion functions as its agglomeration was detected at the binding sites between the bacterium and in the space between the bacterium and gastric cells. Similarly, the immunogold assay of HP0953 protein in the whole bacterium revealed that the protein was primarily located in the bacterial coat or glycocalyx, which is consistent with the finding reported by Smithet al[11], who investigated the proteins secreted byH. pyloriand found that HP0953 protein was a component of this set of proteins.
Furthermore, we alteredHP0953to obtain a mutant strain. Unfortunately, we were unable to propagate the resultantHP0953mutant clone, but we are continuing with our efforts to modify the methodology to obtain a mutant strain that can be successfully propagated. Further studies are necessary to determine the structure and function of hypothetical proteins ofH. pylori, such as HP0953,which may be associated with the disease pathology.
CONCLUSlON
HP0953 protein exhibited its maximum expression during the first 12 h of infection in a culture of gastric epithelium cells. This finding indicated its location surrounding the infected cells, and the predicted presence of prenylation and myristoylation sites suggests that HP0953 is a relevant protein during the bacterium’s colonization phase of the gastric epithelium. HP0953 was also found to be strikingly similar to an exotoxin, possibly associating it with the development of gastric cancer.
ARTlCLE HlGHLlGHTS

ACKNOWLEDGEMENTS
The authors would like to acknowledge Alejandra S Rodríguez for her skillful technical assistance.
FOOTNOTES
Author contributions:Arteaga-Resendiz NK, Rodea GE, Olivares-Cervantes AL, and Colín C performed the experiments, and acquired and analyzed data; Arteaga-Resendiz NK, Rodea GE, Colín C, Olivares-Cervantes AL,Aguilar-Rodea P, Ribas-Aparicio RM, López-Villegas EO, Olivares-Trejo JJ, and Velázquez-Guadarrama N interpreted the data; Reyes-López A and Arteaga-Resendiz NK statistical data analyzed; Rodea GE, Ribas-Aparicio RM, Mendoza-Elizalde S, Salazar García M and Velázquez-Guadarrama N wrote the manuscript; and all authors approved the final version of the article.
Supported bythe Federal Funds, HIM/2009/037. SSA851 and HIM / 2014/012. SSA 1098; the grant from Secretaría de Investigación y Posgrado, SIP 20161878; and the Instituto Politécnico Nacional by Consejo Nacional de Ciencia y Tecnología, CB-222180.
lnstitutional review board statement:The study was reviewed and approved by the Ethics, Biosafety and Scientific committees at the Health Institute approved the experiment (HIM/2009/037. SSA851 and HIM/2014/012. SSA 1098).
lnstitutional animal care and use committee statement:Animal care was performed under national and institutional policies (NOM-062-ZOO-1999).
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to them.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Mexico
ORClD number:Nancy K Arteaga-Resendiz 0000-0003-0777-4212; Gerardo E Rodea 0000-0003-1165-0511; Rosa María Ribas-Aparicio 0000-0002-1141-2794; Alma L Olivares-Cervantes 0000-0001-8209-4896; Juan Arturo Castelán-Ⅴega 0000-0003-1102-2101; José de Jesús Olivares-Trejo 0000-0002-4225-4493; Sandra Mendoza-Elizalde 0000-0002-0255-4125; Edgar O López-Ⅴillegas 0000-0002-9995-2145; Christian Colín 0000-0002-8796-9978; Pamela Aguilar-Rodea 0000-0002-1522-2138;Alfonso Reyes-López 0000-0002-6249-3678; Marcela Salazar García 0000-0001-5318-8148; Norma Ⅴelázquez-Guadarrama 0000-0002-3620-7260.
Corresponding Author's Membership in Professional Societies:Hospital Infantil de México Federico Gómez, 11217.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Gastroenterology2022年29期
World Journal of Gastroenterology2022年29期
- World Journal of Gastroenterology的其它文章
- Mechanistic and functional extrapolation of SET and MYND domain-containing protein 2 to pancreatic cancer
- Clinical challenge for gastroenterologists-Gastrointestinal manifestations of systemic mastocytosis: A comprehensive review
- Structural changes of proteins in liver cirrhosis and consequential changes in their function
- Epidemiologic and socioeconomic factors impacting hepatitis B virus and related hepatocellular carcinoma
- Enhanced endoscopic ultrasound imaging for pancreatic lesions: The road to artificial intelligence
- Qingyi decoction attenuates intestinal epithelial cell injury via the calcineurin/nuclear factor of activated T-cells pathway