
Clinical challenge for gastroenterologists-Gastrointestinal manifestations of systemic mastocytosis: A comprehensive review
2022-08-12 08:10AlessandraElveviElenaMariaElliMartinaLucaMikiScaravaglioFabioPagniStefanoCeolaLauraRattiPietroInvernizziSaraMassironi
World Journal of Gastroenterology 2022年29期
Alessandra Elvevi, Elena Maria Elli,Martina Luca,Miki Scaravaglio, Fabio Pagni, Stefano Ceola Laura Ratti,Pietro Invernizzi,Sara Massironi
Abstract Mastocytosis is a rare and heterogeneous disease characterized by various clinical and biological features that affect different prognoses and treatments. The disease is usually divided into 2 principal categories: cutaneous and systemic disease(SM). Clinical features can be related to mast cell (MC) mediator release or pathological MC infiltration. SM is a disease often hard to identify, and the diagnosis is based on clinical, biological, histological, and molecular criteria with different specialists involved in the patient’s clinical work-up. Among all manifestations of the disease, gastrointestinal (GI) symptoms are common, being present in 14%-85% of patients, and can significantly impair the quality of life.Here we review the data regarding GI involvement in SM, in terms of clinical presentations, histological and endoscopic features, the pathogenesis of GI symptoms, and their treatment.
Key Words: Systemic mastocytosis; Gastrointestinal involvement; Gastrointestinal symptoms
lNTRODUCTlON
Mastocytosis is a rare type of myeloid neoplasm characterized by clonal expansion and accumulation of morphologically and immune-phenotypically abnormal mast cells (MCs) in one or more organ systems[1]. Clinical presentation in mastocytosis patients is heterogeneous[1,2]. Cutaneous mastocytosis (CM) is the skin-limited presentation; it is most common in children, who experience spontaneous regression of skin lesions during their growth. Systemic mastocytosis (SM) is, on the contrary, a condition of MCs proliferation associated with extra-cutaneous involvement, and it is more frequent in adult patients.Neoplastic MCs form focal and/or diffuse infiltrates in various internal organs, including the bone marrow (BM), spleen, liver, and gastrointestinal (GI) tract[2-4]. Regardless of the type of SM, the BM is involved in virtually all patients.
In 2016, the World Health Organization (WHO) classification of SM identified 5 clinical variants with different prognoses: indolent SM (ISM), smoldering SM (SSM), aggressive SM (ASM), SM with associated hematological neoplasm (SM-AHN), and mast cell leukemia (MCL)[5,6]. Advanced SM(ASM, SM-AHN, and MCL) is generally associated with a poor outcome, whereas ISM patients usually have a comparable life expectancy in comparison with the general population[1,2].
In recent years, knowledge of the pathophysiology of mastocytosis has improved. Particularly,research has found that a recurrent activating D816V KIT mutation, located in the phosphor-transferase domain (PTD) of the receptor, is detectable in more than 80% of adult patients with SM[7,8]. Further genetic alterations, usually involved in myeloid malignances pathogenesis, are often found in advanced forms of SM. These mutations are associated with aggressive disease and poor prognosis[9].
For many years, mastocytosis has been considered an orphan disease, and this might be an explanation for the difficulty in estimating its prevalence. The reported prevalence of SM of 9.56 per 100000 persons[10] is probably underestimated; in the last few years, improved knowledge of the disease and availability of diagnostic tests suggested a higher prevalence, with the newer prevalence reported to be 21 per 100000 inhabitants in the adult population[11].
Mastocytosis diagnosis is based on the identification of atypical MCs in the affected tissue, according to the well-established WHO criteria, based on morphological, histological, cytofluorimetric, and molecular features[5,6] (Table 1). When the major and at least one minor SM criterion or three minor SM criteria are detected, the diagnosis SM is established. SM patients can be further sub-classified depending on the presence of B (organ involvement without organ failure) or C (organ involvement with organ dysfunction) findings, defining MCs burden or disease aggressiveness, respectively[1,5,6](Table 2).
The severity of the symptoms associated with mastocytosis may vary from mild to life-threatening[2]. In general, symptoms occurring in SM are mainly due to the release of chemical mediators from the MCs, such as histamine, heparin, and eosinophil chemotactic factor, and thus produce symptoms associated with an allergic reaction, although a true allergic trigger may not be identified. In ASM,where MCs infiltration occurs, a decrease in blood cells (cytopenia), break-down of bones (osteolysis),swelling of the lymph nodes (lymphadenopathy), swelling of the liver (hepatomegaly), impaired liver function, ascites or portal hypertension, and malabsorption may occur[1,12,13].
Symptom-directed treatment should be considered in all SM patients[2]. Supportive therapies for SM are directed at MC’s degranulation symptoms [histamine-receptors 1 (H1) and 2 (H2) antagonists,proton pump inhibitors (PPI), leukotriene antagonist, sodium cromolyn, corticosteroid], symptomatic skin disease (Psolaren plus ultraviolet A photo-chemotherapy, corticosteroids), and/or osteopenia/osteoporosis/pathologic fractures (bisphosphonate, cytokine/ immunomodulatory drugs)[2,14,15]. Treatment goals for ISM patients are primarily directed towards anaphylaxis prevention andsymptom control/osteoporosis treatment. Patients with advanced SM frequently need MCs cytoreductive therapy to ameliorate disease-related organ dysfunction[2]. High response rates have been seen with small-molecules inhibitors that target mutant-KIT, including midostaurin (Food and Drug Administration and European Medicine Agency approved) or avapritinib (Food and Drug Administration approved)[2,16-19].

Table 1 Systemic mastocytosis criteria according to World Health Organization 2016
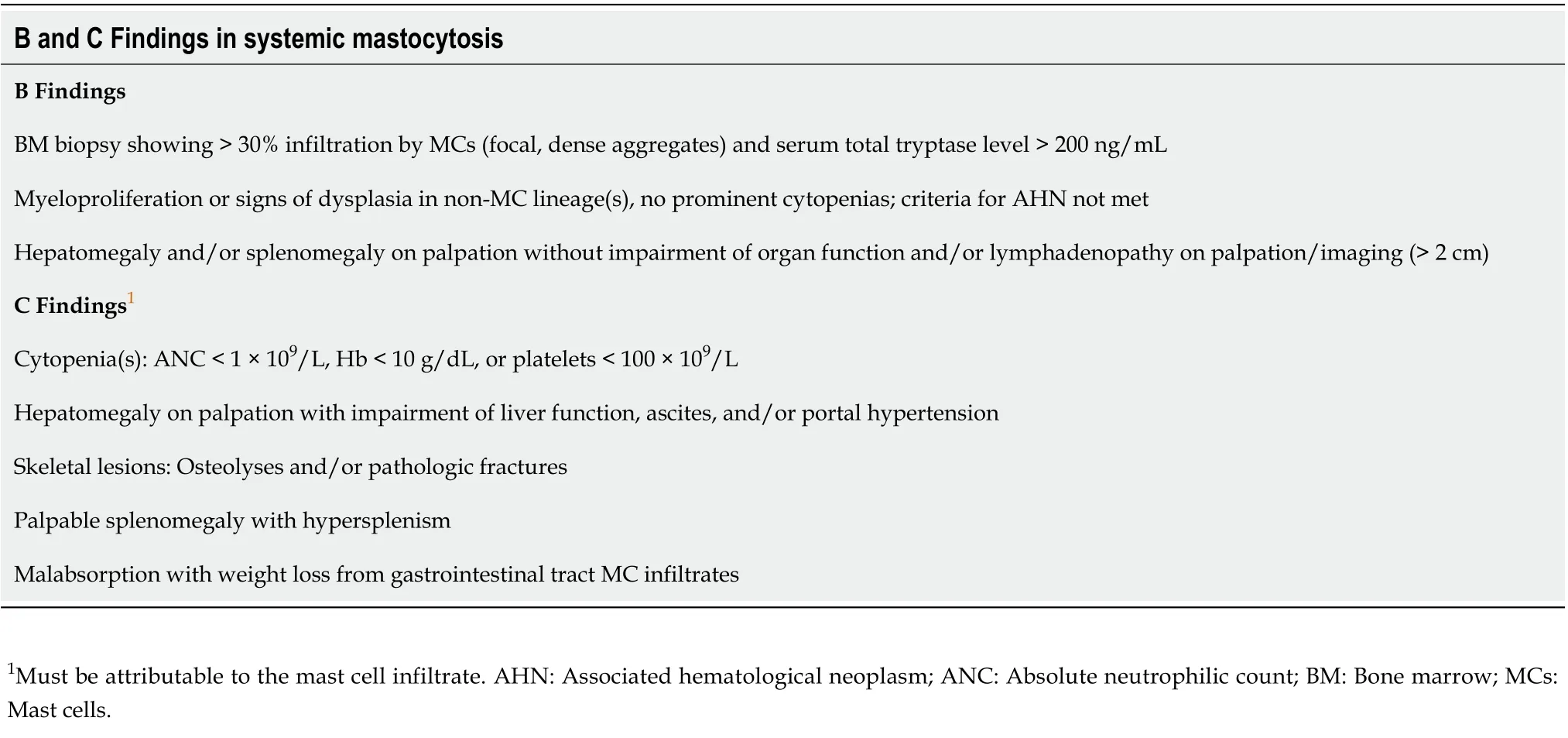
Table 2 B and C Findings in systemic mastocytosis
In this review, we sought to focus on clinical, pathophysiological, histological, and therapeutic aspects of GI tract involvement in SM.
Gl CLlNlCAL MANlFESTATlONS OF SM
GI involvement in the clinical presentation of SM has been reported to vary from 15% to 85%. The wide range of variability is probably due to variable definitions of GI symptoms and differences in the attention on GI involvement in the case series available in the literature[20].However, among all clinical manifestations, GI symptoms appear to be second in frequency only to itching[21] and are the main culprits of chronic disorders being a major determinant of quality of life (QoL)[20-22].
GI symptoms can be varied and may be independent of a clear GI involvement with mastocytes’infiltration[23].In addition, GI manifestations may occur in the absence of typical cutaneous manifestations, with GI dysfunction being observed in both cutaneous mastocytosis and SM[24].Although the gastro-enteric tract involvement has been described in all SM variants, both the frequency and the type of GI symptoms vary according to the SM subtype.
GI symptoms associated with SM are often non-specific and mimic common conditions, such as irritable bowel syndrome, inflammatory bowel disease, or celiac disease, with recurrent abdominal pain,nausea, vomiting, gastro-esophageal reflux disease, diarrhea, and bloating, until more severe pictures characterized by long-standing, persistent, and intractable diarrhea with malabsorption occurs[23].Also, peptic ulcer disease represents a GI manifestation of SM, causing gastro-duodenal ulceration in up to 50% of cases[25], with possible severe GI bleeding sustained by the large amount of histamine released by mast cells (Table 3).
PATHOPHYSlOLOGY OF Gl lNVOLVEMENT
From the pathophysiological point of view, these symptoms are caused by several mechanisms.Activation of MCs causes alteration in epithelial and neuromuscular function. MCs represent 2% of the cells present on the intestinal mucosa of the GI tract[26]: They are responsible for a series of regulatory functions concerning the barrier functions and vascular mucosal permeability, regulating the secretion of GI hormones, modulating peristalsis and the pain perception threshold, and regulating the endothelial barrier and chemotaxis of immune cells. They, therefore, intervene in the regulation of the inflammatory response and, as recently described, they drive the immune response by attracting both granulocytes and lymphocytes and triggering innate defense mechanisms, such as enhanced epithelial secretion, peristalsis, and alarm programs of the enteric nervous system[26,27].
Activated MCs release various inflammatory mediators, which determine intestinal motility alteration, visceral sensitivity alteration, and mucosal and epithelial gut barrier function alteration.Furthermore, MCs inflammatory mediators determine hypersecretion and increased motility, causing diarrhea and abdominal pain. In the more advanced and aggressive stages, there is a direct MC infiltration of the GI tract and the pathogenic mechanisms change. The GI tract can be involved in any phase of SM and the pathophysiological mechanisms are different. As well as reported for other extra-hematological manifestations of SM, in the initial and indolent forms of SM, the gut is a target of mediators released by MCs and functional disorders are more expressed, with mediator-related functional diarrhea, abdominal pain, and motor dyspepsia.
Thereafter, with the massive and persistent mediators’ release, organic lesions appear, such as peptic disease. The main mediator released by MCs is histamine which exerts different effects on both the upper and lower gastrointestinal tract. Histamine interacts with H2 receptors, promoting the secretion of hydrochloric acid, resulting in gastroesophageal reflux disease, gastritis, and peptic ulcer.
The peptic ulcer due to the increase in histamine-related acid secretion is a condition not rarely observed in SM, and it is largely underestimated: It is reported in up to 40% of patients with SM. It does not correlate with circulating markers, in particular with circulating histamine; therefore, it has to be hypothesized with a low threshold of suspicion and then investigated with gastroscopy. Moreover,peptic ulcer bleeding in MS is also reported in up to 11% of patients[25]. Histamine also interacts with the H1 receptors, regulating the gut smooth muscle contraction, causing impaired peristalsis and motility disorders, clinically resulting in the dysmotility-like dyspepsia and functional diarrhea observed in patients in the early stages of SM.
Besides the GI effects of histamine, other transmitters are released in the course of SM, including prostaglandins and serotonin, which have various effects, including effects mainly on the neuromuscular junction of the GI tract, with consequently impaired motility and visceral hypersensitivity. Again, patients present with nausea, vomiting, and abdominal pain worsened by the reduction of the pain threshold. In the course of mastocytosis, diarrhea is often associated with flushing.Therefore, is important to distinguish “wet flushing”, associated with profuse sweating, and “dry flushing”, without sweating (Table 3). In SM, dry flushing is observed, whereas “wet flushing” is more related to physiological or para-physiological causes.
In the more advanced and aggressive stages of MS, the manifestations are mainly related to the gut MC infiltration of the GI wall, which impairs both the barrier functions and the absorbing functions, and the pathogenic mechanisms of diarrhea change from a functional to organic diarrhea. As the disease progresses, the clinical picture evolves into malabsorption, progressive caloric-protein malnutrition,vitamin deficiency, deterioration of the patient's condition, worsening of osteoporosis (already present in these patients), and finally anasarca, due to a complete impaired gut absorption and barrier function.
Mast cell mediators-related GI abnormalities
The release of a great amount of histamine and other peptides contained in MC granules has been implicated in the pathogenesis of a variety of functional GI manifestations in SM, including diarrhea,bloating, nausea, vomiting, abdominal pain, peptic ulcer disease, and related GI bleeding.
A recent multicenter French study analyzing a cohort of 83 patients affected by SM has demonstrated that GI symptoms are more frequent in SM patients compared to matched healthy subjects. Moreover,SM is correlated with a significantly greater risk of developing peptic ulcer disease, especially duodenal ulcers[28]. Diarrhea, bloating and abdominal pain have been reported as the most frequent SMassociated GI disturbances in several studies[20,22,25,29].
Abdominal pain has been described in different case series with a wide range of frequency from 12%to 100%. Of note, Cherner and colleagues[25] identified two different types of abdominal pain. The majority of SM patients suffered from an abdominal pain of dyspeptic nature, defined as an epigastric pain responsive to antacids and histamine type 2 receptor antagonists and with a high incidence of peptic disease on upper gastrointestinal endoscopic examination. A minority of patients complained ofnon-dyspeptic pain associated with bloating that does not benefit from histamine 2 receptor antagonists and antacids and was associated with normal endoscopic investigations. While the pathogenesis of the first pain type may be easily correlated to increased acid secretion, the non-dyspeptic pain type is more likely related to intestinal motility disturbances and a lower pain threshold caused not only by an increased release of histamine but also by other mediators, such as serotonin, prostaglandins, and neuropeptides.

Table 3 Differential diagnosis between systemic mastocytosis and gastrointestinal diseases
Functional diarrhea has been reported in several series of SM patients with a range from 14% to 100%.The presence of diarrhea has been described as an episodic increase in bowel movements with subsequent emission of liquid stool that may or may not be associated with a fecal weight > 200 g/d[20]. It may be related to increased gastric acid secretion with mechanisms similar to Zollinger Ellison syndrome and/or to increased bowel motility induced by prostaglandin D2 secretion, as there is evidence of cases of diarrhea responsive to aspirin administration[30]. In contrast, the presence of steatorrhea has been correlated with a condition of malabsorption characteristic of advanced mastocytosis with extensive mucosal and submucosal infiltration with MCs.
Nausea and vomiting have been described less frequently and usually as components of syndromic disorders that also include palpitations, flushing, abdominal cramps, and diarrhea, generally exacerbated by the ingestion of certain substances, such as alcohol and medications, or contact with inflammatory mediators, which stimulate MCs degranulation. The absence of specific colonic symptoms does not mean that the colon or rectum are not involved. In particular, rectal manometric studies show a lower distention threshold to induce pain or rectal urgency; patients complain of reduced rectal compliance or overactive rectal contractility.
Peptic ulcer disease
One of the most common mediator-related clinical signs, which over time can generate organic lesions and GI organic damage, is related to gastric acid hypersecretion secondary to the hyper-histaminic state,which leads to dyspeptic pain and digestive ulcer disease.
A prospective study shows that peptic ulcers in SM are common, present in up to 50% of patients[25]. It is usually symptomatic and associated with gastric acid hypersecretion and elevated basal acid output (BAO)[31]. In some patients, BAO can reach the Zollinger-Ellison range, so that may even cause the signs and symptoms of the syndrome itself[20]. Of interest, a particular form of CM, the so-calledtelangiectasia macularis eruptiva perstans,also known as paucicellular mastocytosis, has been associated with peptic giant gastro-duodenal ulcers[32]. Other acid-related complications have been reported, such as ulcer perforation and severe esophagitis. Moreover, up to 11% of SM-affected patients will manifest peptic ulcer bleeding during the disease course. However, so far there has been no evidence of a consistent relationship between the level of serum histamine and the presence of ulcer disease or gastric acid output. However, despite the increased circulating histamine level, many patients have normal acid secretion and in some cases, even achlorhydria has been observed[31]. The variability in the response to circulating histamine may relate to partial inactivity of circulating histamine, differences between circulating histamine and gastric tissue histamine, and the intervention of unknown cofactors in the stimulation of oxyntic cells, as well as the theory of possible desensitization of parietal cells that can justify the rare cases of complete achlorhydria[20].
Mast cell infiltration-related GI abnormalities
In patients with advanced SM, the B and C findings (Table 2) become prevalent to functional GI symptoms. B GI findings indicate organ involvement without organ dysfunction, while C GI-findings refer to the advanced stage where both organ infiltration and dysfunction coexist, being associated with a poor prognosis[6]. They include malabsorption, weight loss, and organomegaly (spleen, liver) until full-blown organ dysfunction leads to hepatic failure and ascites.
Malabsorption based on quantitative stool fat analysis, D-xylose, and Schilling tests has been found to occur in one-third of SM patients and is usually mild[32]. Thus, it is important to consider mastocytosis in the diagnostic work-up of chronic and intractable diarrhea, especially when the endoscopic presentation seems to be suggestive. Both the small and the large intestine can undergo permanent alterations, which contribute to the genesis of diarrhea and malabsorption. Over time, patients could develop real intestinal malabsorption syndrome with vitamins A and D deficiency, overt osteomalacia and osteoporosis, protein-energy malnutrition, and anasarca. The whole gastrointestinal tract may be involved in SM with varying degrees.
Liver involvement in SM can range from an abnormal liver function test alone to hepatomegaly due to MC and eosinophil infiltrates in portal triads. Initial fibrosis can evolve in cirrhosis with worsening portal hypertension sustained by simultaneous splenomegaly, with or without hypersplenism, and ultimately ascites and upper digestive bleeding from esophageal varices. Of note, mastocytosis is one of the causes of non-cirrhotic portal hypertension[33],sustained by pre-sinusoidal and sinusoidal mechanisms through the infiltration of inflammatory mast cells within the portal vein and obstruction of the sinusoids[34]. In this setting, we can also observe vascular alterations, like portal hypertension,veno-occlusive disease (8%), and Budd-Chiari syndrome.
No studies have identified how many patients with SM develop portal hypertension, but in real medical practice, clinicians have to keep in mind that it can occur without associated advanced liver disease, further increasing the risk of upper GI bleeding. A study of 24 cases of SM involving the GI tract found only 3 patients with liver involvement; however, liver involvement correlates with more aggressive disease[23]. Moreover, as liver failure occurs, it contributes to worsening malnutrition and hypoalbuminemia that are already determined by intestinal insufficiency.
ENDOSCOPlC FlNDlNGS lN SM
Even though no remarkable endoscopic findings could be found[4], there are several endoscopic pictures described in SM, and endoscopic findings could be secondary both to MC mediator release and to MC infiltration in the GI wall or liver.
Upper gastrointestinal endoscopy
At upper GI endoscopy, peptic disease (esophagitis, esophageal strictures, peptic ulcer) is one of the most frequent findings in patients with SM[21].
According to the literature, upper GI involvement could be found in up to 40% of SM patients undergoing endoscopy[25] and may be sustained by chlorhydric acid hypersecretion. However, its pathogenesis is still uncertain: Peptic disease is thought to be secondary to gastric acid hypersecretion sustained by elevated histamine plasma levels, but data from literature are controversial: Hypersecretion-n of gastric acid does not seem to be consistently related to the level of serum histamine levels measured. It may, however, be possible that a relationship between tissue concentration of histamine and GI manifestations can exist[25].
While esophagitis and, when untreated, esophageal strictures are thought to be secondary to mediator release, esophageal varices could be accounted for SM endoscopic findings when liver infiltration is present; this infiltration determines intrahepatic portal hypertension with esophageal and gastric varices development[33-35].
Exploration of the stomach and duodenum could show urticaria-like lesions in the antrum of the stomach, diffuse telangiectasia, multiple erosions of gastric and/or duodenal mucosa (Figure 1) and peptic ulcer; these findings are thought to be secondary to MCs mediators’ release (i.e.,histamine,serotonin, prostaglandins, and neurotensin). When atypical mastocyte infiltration occurs, nodular mucosal lesion of the stomach and/or duodenum can be found[21,36,37].
Small bowel endoscopy and colonoscopy
Small bowel, as well as other digestive tract components, could be involved by SM. From the review of literature, the most common findings during small bowel exploration are diffuse telangiectasia,thickened jejunal folds, scalloped folds, and diffuse nodularity[21,36]. As for the upper GI tract, diffuse telangiectasia and thickened jejunal folds are thought to be secondary to MC mediator release, while scalloped folds and diffuse nodularity are thought to be secondary to MC infiltration in the GI wall[21].
Colonoscopy in patients with SM can reveal diffuse telangiectasia and/or urticarial lesion, patchy mucosal erythema, as well as mucosal edema, mucosal friability, loss of vascular patterns, and diffuse nodularity. Moreover, adding indigo carmine dye spraying could be useful to enhance yellowish-white polypoid lesions and yellowish-white nodular or granular mucosal lesions[36,38,39].
As for the other GI tracts, mucosal erythema and edema, mucosal friability are thought to be secondary to MC mediator release, while loss of vascular patterns and nodular and /or polypoid lesions are thought to be secondary to MCs infiltration into the bowel wall[21]. Unfortunately, none of these endoscopic signs is specific for GI involvement of SM; hence, biopsy specimens of the GI wall should always be performed in order to have histological diagnostic confirmation of GI involvement.
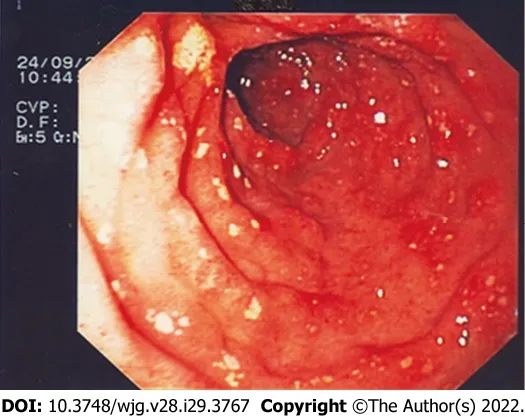
Figure 1 Endoscopic pictures of second portion of the duodenum. The mucosa appears to be erythematous, hyperemic, and covered with diffuse erosions.
HlSTOPATHOLOGlCAL ASPECT lN SM
Mast cells (MCs) are unique granulated cells (approximately 20 μm) that are absent in peripheral blood.They developin situfrom CD34-positive or c-Kit- positive progenitor cells[40]. MCs are distributed throughout many tissues, including the gastrointestinal mucosa. They enable an immune function in response to a diversity of exogenous substances in the gut lumen[41].
Within the gut, two subpopulations can be distinguished: the mucosal reactive MCs, which may dramatically increase in response to immune stimuli and are dependent on T lymphocytes[42,43], and the submucosal MCs, which can act in the regulation of intestinal permeability, secretion, peristalsis,nociception, immunity, and angiogenesis[44]. The histologic appearances of the GI mucosa in SM patients are incompletely described[45]. Results of mucosal mast cell quantitation have similarly been mixed with some studies reporting increased, decreased, and normal numbers of mucosal mast cells in patients with SM compared with controls[46].
The mean normal value of MCs count in normal GI mucosa is yet to be validated in large patient cohorts; most studies consider a cutoff of 20 MCs per high-power field within the lamina propria, this value being 2 standard deviations above that found in the general population[47,48].As MC infiltrate can be focal and mild, the diagnosis of mastocytosis in GI mucosal biopsies can be challenging; hence,multiple biopsies are necessary in order to improve diagnostic yield[28].
In almost all cases of SM involving the GI tract, the number of intramucosal MCs is substantially increased[23], and MCs group together forming pathognomonic compact micronodular or band-like infiltrate. Tiny clusters of cells, with a predominant sub-epithelial distribution, appear in different shapes, varying from rounded cells with a moderate amount of pale and eosinophilic cytoplasm to ovoid and spindle-shaped cells with scant cytoplasm. Those diagnostic infiltrates may be small but should consist at least of 15 clusterings[23].
Hematoxylin and eosin staining are not a definitive method for MCs detection, hence special staining is required, such as toluidine blue staining and immuno-histo-chemical panels using tryptase, CD117,and CD25[49]. Comparison between these techniques confirmed a stem cell factor receptor kit (testing for c-kit and CD117) is the most accurate technique[50]. CD117 is expressed on all types of MCs independent off maturation and activation status; in SM, c-Kit is often expressed in MCs in a mutated and constitutively activated form. In these patients, MCs aberrantly display CD25, a diagnostic marker of neoplastic MCs in all SM variants[51]. Therefore, CD117 is sensitive, but not specific for MCs; tryptase is less sensitive but more specific. Both CD117 and tryptase are not diagnostic of the neoplastic nature,whereas CD25 expression is considered a hallmark of MCs atypia[4] (Figure 2).
SM may involve other GI organs like the liver. The main site of liver involvement is the portal tract,where MCs may form groups of dense infiltrates within the fibrotic portal triads; because MCs are normally absent in the sinusoid, the presence of scattered MCs in liver sinusoids is also consistent with SM (Figure 3).
TREATMENT OF Gl SYMPTOMS
Symptom-directed treatments should be considered in all SM patients and are directed at MC degranulation symptoms (i.e.nausea, vomiting, abdominal pain, peptic disease, diarrhea)[2].

Figure 2 Spectrum of histologic findings in patients with intestinal involvement of systemic mastocytosis. A: A mild initial finding with mast cells arranged in sheets and micro aggregates (hematoxylin-eosin, × 10). A heavy eosinophil infiltrate frequently dominates the picture. Crypt architectural distortion,without any other evidence of inflammatory bowel disease. Scattered lymphocytes and plasma cells often accompanied the mast cell and eosinophil infiltrates; B-C:The mast cells show expression of c-kit (B; CD117, × 2) and tryptase (C; × 10); D: CD25 positive cells in a clearcut and spread duodenal mastocytosis (CD25, × 10).
As first step, patients must recognize and avoid general and specific individual factors that trigger symptoms. These can commonly include physical factors such as heat, temperature changes, some forms of exercise, emotions, stress, sleep deprivation; diagnostic and therapeutic agents (such as opiates,NSAIDs, succinylcholine, and agents with tetrahydroisoquinoline, atracuronium, rocuronium, and quinolones), foods, alcohol and Hymenoptera venoms[52].
Different symptomatic drugs have been proposed for the control of GI symptoms: H2- antagonists (i.e.,Famotidine 10 mg BID, Cimetidine 400 mg BID) are the first-line therapy[2]. They act as inverse agonists that combine with and stabilize the inactive conformation of the histamine-receptor, shifting the equilibrium toward the inactive state; in a concentration-dependent manner, they inhibit MC activation and histamine release. However, although,the mechanisms involved have not yet been delineated fully, downregulation of intracellular calcium ion accumulation seems to play a role[53].Inactivation of histamine receptors reduces the histamine effect along the GI tract, hence reducing histamine-related symptoms (i.e.,diarrhea, abdominal pain, peptic disease).
When histamine-receptor blockers are poorly effective or ineffective, proton pump inhibitors (PPIs,i.e.,Omeprazole 20 mg/d, Rabeprazole 20 mg/d, Pantoprazole 40 mg/d) represent second-line therapy[2]. Hydrochloric acid is secreted by parietal cells in the oxyntic mucosa that lines the body and the fundus of the stomach as a response to different mediators (i.e.,histamine, gastrin, acetylcholine), that activate H+/K+ ATP-ase (i.e.,the proton pump) that pumps hydrogen ions (protons) into the lumen in exchange for potassium ions. PPIs block this final step in gastric acid secretion by blocking H+/K+ ATPase irreversibly. They are remarkably effective in the inhibition of gastric acid secretion[54]. Considering SM as a hypersecretory condition, it is clear that reducing hydrochloric acid secretion is beneficial in SM patients because it reduces peptic symptoms, peptic disease, and/or diarrhea.
Sodium cromolyn (100-200 mg QID 30 minutes before meals and bedtime) represents the third-line therapy when H-2 antagonists and PPIs are ineffective[2]. It works as an inhibitor of histamine release and cell membrane stabilizer: cromolyn sodium caused weak inhibition of histamine release. However,different studies have shown its action on other mediators involved in mast cells' degranulation by inhibiting prostaglandin D2 release and TNFα release from mast cells, hence reducing MC degranulation and mediator release[55]. Reducing mediator release should help reduce GI symptoms.

Figure 3 Frustules of hepatic parenchyma with substantially preserved structure, with trabeculae with 2-cell spinnerets with no significant steatosis, but focal balloon-like degeneration associated with biliary stasis phenomena. A: Biliary spaces with preserved ducts, with ductular regeneration and bilio-hepatocyte metaplasia (hematoxylin-eosin, × 4); B: Some phenomena of hepatitis with aggression of the biliary epithelium are observed (hematoxylin-eosin, × 20). Portal spaces enlarged due to the presence of mixed inflammatory infiltrates, mainly consisting of T lymphocytes (CD3 +) with initial fibrotic expansion and formation of porto-portal bridges; C: In this context, scattered CD117 +, tryptase + cells (× 20) referable to mast cells.
Pardanani[2] has proposed systemic corticosteroid (i.e.prednisone 0.5-1 mg/kg/d starting dose; taper as feasible based on response/tolerance), as the fourth line of therapy for GI symptoms resistant to previous symptomatic therapies. Corticosteroids are potent anti-inflammatory drugs widely used in the treatment of allergic and inflammatory diseases. They also showed an in-vitro inhibition of MCs mediators’ release. Systemic administration of corticosteroids has been used in patients with mastocytosis. While this treatment appears to improve control of some mediator-release symptoms, it has not been proven to reduce MC numbers significantly in patients with mastocytosis[56]. Of note,systemic corticosteroids have been useful in decreasing the malabsorption and ascites in some of these patients[57].
In advanced SM, treatments interfering with MC proliferation and survival are used[2]. GI symptoms secondary to organ infiltration could also benefit from cytoreductive therapies, such as 2-chlorodeoxyadenosine (cladribine or 2-CdA) or interferon-alfa[58]. In young and otherwise healthy ASM patients,allo-SCT is the only option for a sustained response[59]. Promising effects have been seen with the use of targeted therapy on mutated-KIT, which occurs in most SM patients[2]. Midostaurin (PKC412) is the first kit-inhibitor recent approved by the United States Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of Advanced SM[16,17]. It hasin vitroactivity against kinase domain KIT mutants (D816Y and D816V). In the global Phase 2 CPKC412D2201 trial[18],116 patients with advanced SM were enrolled; patients were treated with PKC412 at 100 mg BID. The overall response rate (ORR) per conventional criteria was 60% with 45% having a major response and 15% having a partial response, but it is often not sustained. In particular, concerning GI involvement,reversal of organ damage was reflected by normalization of hypoalbuminemia (58%), improvement in liver function test abnormalities (44%-58%), and/or reversion of weight loss (25%). Patients reported improvement of disease-related symptoms with treatment. Therefore, the most common drug side effects (all grades/grades 3-4) were nausea (79%/6%), vomiting (66%/6%), diarrhea (54%/3%), and fatigue (28%/9%), in addition to hematological toxicity, in terms of grade 3-4 of neutropenia, anemia,and thrombocytopenia, occurring in 24%, 41%, and 29% of patients, respectively.
Avapritinib[19], a KIT and PDGFRA inhibitor, showed promising results; other medications potentially effective for SM patients are under investigation[2]. Further optimization of treatment with tyrosine-kinase inhibitors in order to improve patients' quality of life is still needed.
CONCLUSlON
Mastocytosis is a rare and heterogeneous disease characterized by various biological and clinical features with different prognosis and treatments. The disease is usually divided into 2 principal categories: CM and SM. Clinical features can be related to MCs mediators release or pathological MCs infiltration. The diagnosis of SM is based on clinical, biological, histological, and molecular criteria,according to WHO 2016 classification. A proper patient's workup requires a multidisciplinary approach including gastroenterologists, endoscopists, hematologists, and pathologists. Among all manifestations of the disease, GI symptoms are common, being present in 15%-85% of the patients and can significantly impair the quality of life. Here, we review the data regarding GI involvement in SM, in terms of clinical presentations, histological features, pathogenesis of GI symptoms and their treatment. The most frequent GI symptoms are abdominal pain, diarrhea, nausea, and vomiting. GI lesions may involve all the digestive tract, from the esophagus to the rectum. The histological diagnosis of GI involvement is difficult. The treatment of GI symptoms aims to prevent and limit MCs degranulation and/or its consequences and more rarely to control tumoral mast cells infiltration. The diagnosis of mastocytosis should be considered in the case of unexplained severe GI disorders.
ACKNOWLEDGEMENTS
Massironi S and Invernizzi P are members of the European Reference Network on Hepatological Diseases (ERN RARE LIVER) and they thank AMAF Monza Onlus and AIRCS.
FOOTNOTES
Author contributions:Elvevi A, Elli EM, and Massironi S designed the study; Elvevi A, Elli EM, Lucà M, and Scaravaglio M wrote the first draft of the paper; Pagni F, Ceola S, and Ratti L revised the paper and wrote the final version; Massironi S and Invernizzi P reviewed the paper for important intellectual content; all authors have read and approved the final manuscript.
Conflict-of-interest statement:All authors report no relevant conflict of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Italy
ORClD number:Alessandra Elvevi 0000-0001-9841-2051; Elena Maria Elli 0000-0001-5576-5412; Martina Lucà 0000-0003-3787-6973; Miki Scaravaglio 0000-0003-2914-4908; Fabio Pagni 0000-0001-9625-1637; Stefano Ceola 0000-0002-5815-987X;Laura Ratti 0000-0003-0198-6433; Pietro Invernizzi 0000-0003-3262-1998; Sara Massironi 0000-0003-3214-8192.
S-Editor:Wu YXJ
L-Editor:Filipodia
P-Editor:Wu YXJ
 World Journal of Gastroenterology2022年29期
World Journal of Gastroenterology2022年29期
- World Journal of Gastroenterology的其它文章
- Mechanistic and functional extrapolation of SET and MYND domain-containing protein 2 to pancreatic cancer
- Structural changes of proteins in liver cirrhosis and consequential changes in their function
- Epidemiologic and socioeconomic factors impacting hepatitis B virus and related hepatocellular carcinoma
- Enhanced endoscopic ultrasound imaging for pancreatic lesions: The road to artificial intelligence
- Qingyi decoction attenuates intestinal epithelial cell injury via the calcineurin/nuclear factor of activated T-cells pathway
- High-fat diet aggravates colitis via mesenteric adipose tissue derived exosome metastasis-associated lung adenocarcinoma transcript 1