
界面聚合法制备用于脱氮提纯CH4的N2优先渗透ZIF-90/聚酰胺混合基质膜
2022-08-10 09:49于喆淼王志生梦龙邢广宇王纪孝
化工学报 2022年7期
于喆淼,王志,生梦龙,邢广宇,王纪孝
(天津大学化工学院化学工程研究所,天津 市膜科学与海水淡化技术重点实验室,化学 工程联合国家重点实验室,天津化学化工协同创新中心,天津 300350)
引 言
以甲烷(CH4)为主要成分的天然气是一类蕴藏丰富的低碳能源[1],在国民生产生活中发挥着重要作用。天然气可以分为常规天然气和非常规天然气,常规天然气从常规油气藏中开发并被人们广泛使用,非常规天然气比如煤层气、深盆气、页岩气、油砂岩气,在地下的赋存状态和聚集方式与常规天然气具有明显差异。随着常规天然气勘探开发难度不断加大,能源领域迫切需要研究开发利用非常规天然气[2]。然而,非常规天然气处理难度大,经预处理后无法满足N2含量<4%(体积)的管道输送要求,且N2过量会导致气体传输体积增加,降低天然气的能源效率[3-4],因此开发新技术以实现非常规天然气中N2与CH4的高效分离已成为世界油气工业发展的必然趋势[5]。
膜分离法具有占地面积小、操作弹性大、资金投入少、放大效应不显著等优点,是一种很有潜力的气体分离技术[6-7]。用于分离N2/CH4体系的气体分离膜可以分为CH4优先渗透膜和N2优先渗透膜,相较前者,N2优先渗透膜优势在于分离后CH4产品处于高压侧,压力损失小,有利于后续处理[8]。因此设计和开发用于脱氮提纯甲烷的高性能N2优先渗透膜及膜过程有望产生巨大的经济和社会效益。
由于N2和CH4尺寸上差异很小(图1),且二者与聚合物基质间相互作用差异很小,考虑到N2分子动力学尺寸较小且惰性强,临界温度(TC=126.1 K)低于CH4(TC=190.7 K),因而冷凝性较弱,故提高扩散选择性[10]同时增强筛分作用对于改善氮气优先透过膜的分离性能至关重要。根据制膜材料的不同,目前研究的N2优先渗透的N2/CH4分离膜主要包括无机分子筛膜和有机聚合物膜。分子筛膜可以由SAPO-34、ETS-4、DDR 等沸石分子筛和碳分子筛材料制备[11-13],Wu 等[14]采用蒸汽辅助转化方法合成了薄而高质量的SSZ-13 膜用于分离N2和CH4,测试结果显示,当进料压力为2 bar(1 bar=0.1 MPa)时,该膜N2渗透速率为1.40×10-7mol·m-2·s-1·Pa-1,N2/CH4分离因子为14.8;有机高分子聚合物膜可以由聚酰亚胺类、全氟聚合物及其衍生物和热重排聚合物等具有刚性分子链段的材料制备[10,15-17],Nikiforov 等[18]利用六氟丙烯(HFP)共聚物制膜,该膜N2渗透速率为5.19×10-12mol·m-2·s-1·Pa-1,N2/CH4分离因子为6.2。无机分子筛膜虽然可以得到较高的渗透速率,但其制备过程复杂,成本较高,单位体积内装填的膜面积较小;有机聚合物膜虽然制备成本较低,且单位体积内装填的膜面积较大,但大多数N2优先渗透聚合物膜存在膜中聚合物链段的无序堆积现象,会导致膜孔径不精准、贯通膜的孔数少、传递路径曲折等问题。故现阶段聚合物膜的渗透分离性能难以满足脱氮提纯甲烷的要求。

图1 CH4、N2分子的尺寸与形状[9]Fig.1 Sizes and shapes of CH4 and N2molecule[9]
界面聚合(IP)法操作简单、可控性强,通过调节如反应时间、反应温度、单体及其浓度、添加剂纳米颗粒的粒径等条件,可以调节聚合物链段堆积的致密程度、膜孔径以及气体在膜中传递路径。目前应用该方法制备的兼具高水通量和高脱盐率的薄膜复合反渗透膜,已经实现工业化[19-21],但该方法用于制备气体分离膜的相关报道还比较有限[22-23]。Ali 等[24]利用延长反应时间与提高有机溶液温度的方法改进了IP 工艺,成功制备了高度交联、超高选择性且无缺陷的MPD-TMC 聚酰胺薄膜复合分子筛膜用于H2/CO2分离。实验表征结果显示,延长反应时间有助于使更多MPD 参与反应,提高聚酰胺产率,在一定程度上减少了超薄膜中的缺陷,提升了所制膜性能。
掺入纳米材料对聚酰胺薄膜复合膜进行改性是一种提高聚合物膜性能的有效方法。在聚合物基质中掺杂金属有机框架(metal organic frameworks,MOFs)填料,通过有机-无机复合结构发挥填料的选择性吸附以及尺寸筛分等特殊作用,为气体传输提供传递通道,可提高膜的渗透选择性。在An 等[25]的研究中通过在聚酰胺层内部掺杂ETS-4 分子筛材料制备了薄层纳米复合膜,产生更多的自由体积,提供具有N2选择性的气体传输通道,明显改善了膜的N2/水蒸气分离性能。金属有机骨架ZIF-90 是一种小孔径、高孔隙率的新型材料,其孔径0.35 nm[26],由于CH4、N2分子的尺寸及形态差异其孔道可以允许N2分子通过而不允许CH4分子通过[27]。以ZIF-90 纳米颗粒为填料,其内部微孔可为N2提供高速传输通道,从而提升N2在膜内的渗透速率;同时,其含有的醛基可同聚酰胺链端的氨基交联增加界面相容性,并有助于颗粒在膜内分散。
使用均苯三甲酰氯油相单体和间苯二胺水相单体,首先在界面聚合过程中分别通过调节两相单体浓度,改变反应的条件,加强链间的紧密堆积。再通过在聚合物基质中引入孔道允许N2分子通过而不允许CH4分子通过的纳米级多孔材料ZIF-90,制备混合基质膜。探究了单体浓度及颗粒含量对膜分离性能的影响。
1 实验材料和方法
1.1 材料与试剂
聚砜(PSf)基膜由贵阳时代沃顿科技有限公司提供。间苯二胺(MPD,99%)、三乙胺(TEA,99%)、樟脑磺酸(CSA,99%)、聚乙烯吡咯烷酮(PVP,99%)、四水硝酸锌[Zn(NO3)2·4H2O,99%]来源于阿拉丁试剂(中国)有限公司。咪唑-2-甲醛(ICA,99%)来源于天津希恩思有限公司。均苯三甲酰氯(TMC,99.5%)来源于青岛三力化工有限公司。正庚烷(分析纯)、叔丁醇(分析纯)、甲醇(分析纯)来源于天津市江天化工技术有限公司。
1.2 ZIF-90纳米颗粒的制备
首先配制两种溶液。溶液A:将叔丁醇与H2O以1∶1 的体积比混合(共20 ml)后得到均相溶剂,向其中加入Zn(NO3)2·4H2O(236.25 mg)。溶液B:将去离子水和甘油以1∶1 的体积比混合(共20 ml),向其中加入咪唑-2-甲醛(480.0 mg)和PVP(50.0 mg),在65℃下搅拌溶解。在室温环境中将溶液A添加到冷却至室温后的溶液B 中,剧烈搅拌5 min 后,停止搅拌。置于离心机中以10000 r·min-1的速度离心,再使用甲醇洗涤3 次(每次30 ml),最后得到ZIF-90 材料,并在50℃下真空干燥[28]。
1.3 N2/CH4分离膜的制备
本文所采用的基膜为高孔隙率的PSf超滤膜(平均截留分子量为45000,表面孔隙率约0.87%,表面平均孔径约为11.7 nm)。混合基质复合膜制备过程反应方程式如图2所示。
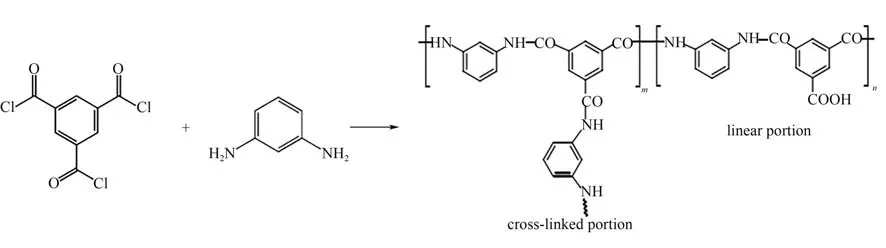
图2 生成聚酰胺膜的化学反应示意图Fig.2 Schematic diagram of chemical reaction for forming polyamide membrane
首先将有机单体TMC 溶解于正庚烷中,充分搅拌1 h 制成油相溶液,再分别取水相单体MPD 溶解于去离子水中,加入三乙胺(TEA)和樟脑磺酸(CSA)制成几份相同的水相溶液。其中两种反应单体分别溶于不互溶的水相和有机相,可在基膜表面通过界面聚合反应生成聚酰胺复合膜;三乙胺(TEA) 和樟脑磺酸(CSA)作为酸吸收剂和pH 缓冲剂加入水相溶液,以中和聚合过程中酰氯基团与氨基反应产生的HCl,防止HCl 与氨基反应生成没有反应活性的氨基盐酸盐,影响成膜过程。另取一定量的纳米颗粒分别加入上述水相溶液或油相溶液中,充分搅拌并超声分散制成分散液。在本研究中,通过改变水相、油相单体浓度以及纳米颗粒添加量等制膜条件来改善膜的性能,制膜条件见表1。

表1 制膜条件Table 1 Membrane preparation conditions
界面聚合法制膜的主要步骤是:将聚砜基膜贴于膜框中,倒入水相溶液浸泡5 min,倒出水相溶液后,利用滚轮将膜表面多余溶液压入基膜的孔内。接下来将膜浸入80℃的有机相溶液中使得油相溶液和膜表面残留的水相溶液充分接触,界面聚合反应5 min,然后立即置于烘箱中(80℃)进行热处理4 min,使聚酰胺层固化从而形成致密的聚酰胺薄膜。最后,将膜转移到恒温恒湿箱中,在设定的温度(30℃)、相对湿度(40%)下干燥24 h。混合基质膜制备过程及结构如图3所示。
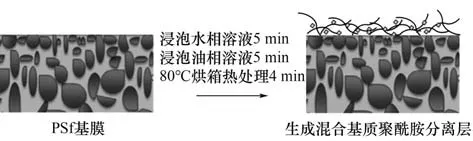
图3 混合基质膜制备及结构示意图Fig.3 Preparation and structure of mixed matrix membrane
此外,Zhang 等[29]研究探索了一种称为“基膜孔堵塞(SMPB)”的现象,指出界面聚合法制备聚酰胺复合膜的过程中由于有机相侵入,在聚醚砜基膜的孔中可以发现聚酰胺结构。Singh 等[30]采用与本研究类似的聚砜基膜,对基膜和薄膜复合膜的聚酰胺层进行ATR-IR 和SEM 表征,证明了孔径较大的基膜(150 nm)比孔径较小的基膜(70 nm)聚酰胺对聚砜基膜孔的渗透量增大,聚砜基膜的孔被聚酰胺堵塞,分离层更薄。以上的研究均证实了聚酰胺膜在生长过程中能够生长到聚砜多孔膜的孔道中。本文中聚酰胺也会生长到聚砜基膜的孔道中,这是由于在制膜过程中,多余的水相溶液被滚轮压入基膜的孔内,因此再加入油相溶液反应时,油相溶液侵入基膜孔导致在基膜中靠近基膜表面的孔道内形成聚酰胺,但是因本研究使用的基膜孔径仅11.7 nm,所以不会形成额外的缺陷。此外,聚酰胺生长到聚砜膜的孔道可以增大层间结合力使制备的复合膜更加稳定,嵌入基膜表面孔中的聚酰胺也有助于提升复合膜的气体筛分性能[31]。
1.4 膜与颗粒的表征
1.4.1 X 射线衍射(XRD) 采用日本理学公司生产的D/MAX-2500型X 射线衍射仪对纳米材料的晶体结构进行表征。将待测纳米材料放置于50℃的真空干燥箱中干燥12 h 后即可检测。扫速设定为5(°)·min-1,范围4°~60°。
1.4.2 衰减全反射-傅里叶变换红外光谱(ATRFTIR) 使用美国Bio-Rad 公司生产的FTS-6000 型ATR-FTIR 分析仪对制备的纳米材料及膜样品进行化学结构分析。将制备的纳米材料及膜样品放入50℃的真空干燥箱中干燥12 h后即可检测。
1.4.3 扫描电子显微镜(SEM)及能量色散X 射线光谱(EDS) 使用美国FEI 公司生产的Nova Nano430型发射扫描电子显微镜对制备的纳米材料及相应的复合膜样品的表面和断面形貌进行表征,并使用仪器配有的能量色散X射线光谱表征样品表面的特征元素。
测试前需要将样品充分干燥,使用导电胶固定在电镜台,最后进行喷金处理。其中断面的制备需要先将分离层与基膜剥离,置于去离子水中润湿后再置于液氮中20 s,取出后迅速折断,再将分离层向外固定于断面台上。
1.5 膜与颗粒的表征
膜的渗透选择性能用课题组自制的气体渗透装置测量。在22℃时,将待测试的膜安装在圆形不锈钢膜池中(膜池橡胶垫圈面积为4.9 cm2,故膜的有效面积取4.9 cm2),以纯N2、纯CH4、混合气N2/CH4(体积比:50/50)作为进料气,He作为吹扫气,在进料侧压力为2~6 bar、渗透侧为常压的条件下进行测试(表压为1~5 bar,吹扫气体的反扩散对数据分析的影响可以忽略不计[32])。扫气出口的流速由肥皂膜流速仪获得,渗透侧气体组分由配有热导检测器的气相色谱仪(HP 7890A)分析。
渗透速率定义为气体渗透通量除以某种组分进料侧和渗透侧间的分压差:

2 实验结果与讨论
2.1 纳米颗粒的表征
如图4 所示,合成的ZIF-90 颗粒形貌如文献报道[26-27],在SEM 图像中观察到典型的高均匀度菱形十二面体形状。ZIF-90 纳米颗粒的平均尺寸约为200 nm。

图4 ZIF-90纳米颗粒的SEM图Fig.4 SEM image of ZIF-90 nanoparticles
图5 为合成的ZIF-90 的XRD 谱图。在10.5°,12.7°,14.5°,16.5°,18.0°,22.3°,26.8°处可观察到明显的特征尖峰,与文献[27]相符,证实样品具有高结晶度。
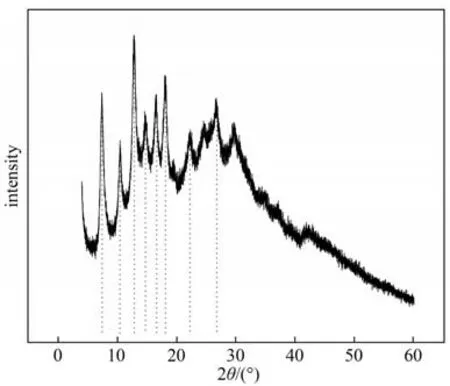
图5 ZIF-90纳米颗粒的XRD谱图Fig.5 XRD patterns of ZIF-90 nanoparticles
图6 中曲线a 为在水-醇溶剂热体系合成的ZIF-90 纳米材料的ATR-FTIR 图,与以N,N-二甲基甲酰胺(N,N-dimethyl-formamide, DMF)为溶剂的溶剂热体系中合成的ZIF-90 具有相同的官能团[27],而1450 cm-1是由咪唑环中N—H 键面内弯曲振动产生的,1400 cm-1处的吸收峰是由制备纳米颗粒所使用的单体中咪唑的C—N 键伸缩振动产生的,因此ATR-FTIR 表征证实样品中存在大量的咪唑和醛基基团,ZIF-90纳米颗粒被成功合成。

图6 ZIF-90纳米颗粒与膜的ATR-FTIR 谱图a―ZIF-90纳米颗粒;b―含0.50 g·L-1纳米颗粒的混合基质复合膜;c―不含纳米颗粒的复合膜;d―PSf基膜Fig.6 ATR-FTIR spectra of ZIF-90 nanoparticles and the membrane a―ZIF-90 nanoparticles;b―mixed matrix composite membrane containing 0.50 g·L-1 nanoparticles;c―polyamide composite membrane;d―PSf substrate
2.2 膜微观结构的表征
图7 显示了掺杂不同含量ZIF-90 颗粒的混合基质膜表面与断面形态。当放大倍数为10000 倍时,图7(b)显示聚砜基膜表面呈现出光滑的多孔结构,经过界面聚合过程后,生成的聚酰胺复合膜表面形貌图7(a)、(d)、(f)、(i)、(l)显示所有聚酰胺复合膜表面都表现出典型的“峰-谷”形态,这是由于界面聚合过程在基膜表面形成了聚酰胺分离层[34]。此外,随着添加量增加,膜表面叶片结构逐渐减少,这是由于分散在水相和有机相反应界面中的纳米颗粒会阻碍MPD 扩散,从而影响了界面聚合过程的效果。锌元素在膜表面EDS 图[图7(g)、(j)、(m)]中显示为白色亮点,锌元素在膜中分布相对均匀,且随着纳米颗粒添加量增加,膜表面锌元素含量增加,故推断制备的纳米颗粒在膜中分布相对均匀。

图7 PSf基膜、M2-T0.1膜、M2-T0.1-Z0.01膜、M2-T0.1-Z0.03膜、M2-T0.1-Z0.065膜的SEM图及表面EDS图Fig.7 SEM images and surface EDS images of PSf substrate,M2-T0.1 membrane,M2-T0.1-Z0.01 membrane,M2-T0.1-Z0.03 membrane,M2-T0.1-Z0.065 membrane
由断面图7(e)、(h)、(k)、(n)可知随添加颗粒浓度升高膜的选择层厚度增加,这是由于纳米颗粒聚集在膜表面,导致了粗糙度增加。此外通过对比图7(a)、(d),可知对聚酰胺膜进行热处理后,膜表面“峰-谷”结构增加程度更加明显,这说明对膜进行热处理固化可对膜结构产生较大影响。对比图7(c)、(e)可以看出,对膜进行热处理可以使膜厚度从约300 nm 增加到400 nm,这是由于热处理使膜内残基进一步交联,增加分离层交联度,聚酰胺分子链增长、链刚性增强,链段堆积空间增大,导致膜厚增加并使膜结构更稳定。
利用ATR-FTIR 表征聚酰胺分离层是否成功合成以及MOF 颗粒是否被成功掺杂在膜中。如图6所示,由于在复合膜的ATR-FTIR 谱图曲线b、c 中并未出现均苯三甲酰氯在1774 cm-1处的酰氯(—COCl)振动特征吸收带[35],这证明了均苯三甲酰氯已在界面聚合过程中充分反应完全,但是谱图中在1659 cm-1处出现了酰胺键中C==O 的伸缩振动特征吸收带,在1540 cm-1处出现了酰胺键中N—H 平面弯曲振动特征吸收带,这两个吸收带的出现表明生成了酰胺键[36]。并且这些在制备出的聚酰胺复合膜中都存在的与聚酰胺官能团相关的特征吸收带,均不存在于聚砜基膜的谱图曲线d 中,这清楚地表明了聚酰胺薄膜经过界面聚合反应在聚砜基膜上形成。此外,对比未经掺杂的聚酰胺复合膜,掺杂纳米颗粒的混合基质膜与纳米颗粒特征衍射峰的位置几乎相同,即可以证明纳米颗粒成功掺入膜中,且颗粒结构没有被聚酰胺破坏。
2.3 单体浓度对膜性能的影响
水相和油相单体的浓度会通过影响界面聚合反应过程,从而影响膜的化学结构和渗透选择性能。因此,本节考察两相单体的浓度对膜渗透选择性能的影响规律。
由于N2和CH4均不与聚酰胺膜发生反应,它们在膜中的传输基于分子筛分机理。如图8 所示,固定TMC 的浓度0.10%,MPD 浓度2.0%时,在纯气测试中,由于N2和CH4的分子动力学直径接近,因此两种气体在膜中的渗透速率相差不大。在纯气测试中,纯N2渗透速率随压力升高而减小,是由于压力的增加会导致膜压实,膜结构更加致密,从而降低膜的N2渗透性。此外,在压力循环测试中将进料压力从最低测试压力升高至最高测试压力,再从最高测试压力降低至最低测试压力,膜的N2渗透速率在同一压力下测得数据基本相同,这表明压实是可逆的,同时说明了膜在测试压力范围内没有缺陷产生。纯CH4压力循环测试也显示渗透速率在同一压力下的测试结果基本相同,但纯CH4渗透速率随压力升高而增加,这是由于相比N2(TC=126.1 K),CH4具有较高的临界温度(TC=190.7 K),因而冷凝性较强,随着压力升高甲烷在膜内溶解度大大增强,导致大量CH4分子进入聚合物链间,聚合物链段本身间的相互作用力减弱,链的灵活性增强,从而气体在膜内的扩散系数增加,CH4分子更易透过。
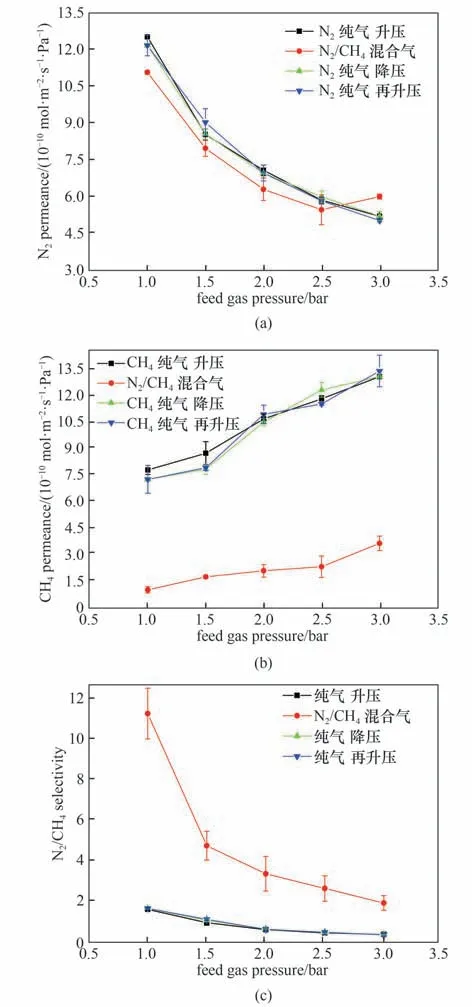
图8 压力对膜气体传递性能的影响[水相中MPD浓度为2.0%,油相中TMC浓度为0.10%,进料气为N2/CH4混合气(体积比:50/50),N2、CH4纯气[>99.999%(体积)],其中纯气为压力循环测试]Fig.8 Effect of feed gas pressure on membrane gas permselectivity[MPD concentration 2.0%,TMC concentration 0.10%,feed gas:N2/CH4(50/50 by volume),pure N2 and CH4 gas(>99.999%by volume),the pure gas test was carried out with pressure cycle]
在N2/CH4混合气测试中,在气体分压1~2.5 bar时,两种气体渗透速率均低于纯气渗透速率,在3 bar时,混合气中N2渗透速率仅略高于纯气,CH4渗透速率仍低于纯气,且在气体分压为1~3 bar时混合气N2/CH4分离因子明显大于理想分离因子,这是由于N2和CH4分子在膜内的传递存在竞争关系,N2分子动力学直径相较于CH4更小,故使用混合气测试时,N2分子更容易进入膜内,而进入孔道内的N2分子会阻碍CH4分子在膜孔道内的传递,因此混合气中的CH4渗透速率比纯CH4低,最终表现为膜的N2/CH4混合气选择性高于纯气。N2/CH4混合气测试中CH4渗透速率均随压力升高而增加,N2渗透速率随压力升高呈现为先减小后增大的趋势,这是由于芳香聚酰胺分离层成膜过程中会产生不同大小的气体传输孔道,包括孔尺寸仅允许N2透过而不允许CH4透过的小孔道,以及孔尺寸允许N2和CH4透过的大孔道,由于CH4冷凝性强,在膜内的溶解作用更强,因而与膜的结合力也强于N2,因此是CH4优先进入大孔道并仅从其中透过,影响了N2在大孔道内的扩散,导致大部分N2都从小孔道中透过。随着压力升高,更多的CH4进入膜,弱化了聚酰胺链段之间的结合力,聚合物链段的灵活性增强,使得CH4更容易透过,从而导致了CH4的渗透速率在测试压力范围内均随压力升高而升高;而对于N2渗透速率,当压力较低即N2分压低于2.5 bar 时,由于随着压力升高膜结构更加致密,抑制了分子尺寸较小的N2从小孔道中渗透,因而此范围内N2渗透速率随压力升高而降低,当压力较高即N2分压高于2.5 bar 时,由于受到更多CH4进入膜对膜结构的影响,聚合物链段的灵活性更大程度增强,导致N2的渗透速率随压力升高而升高,而纯N2渗透速率随压力升高呈现单调降低趋势的另一个原因也是纯N2渗透没有受到CH4的影响。从图8 还可以观察到,在气体的分压为1~3 bar时混合气的N2/CH4分离因子均大于1,即测试压力范围内N2渗透速率均大于CH4,这也证实了N2/CH4的分离主要是扩散选择性主导,而不是溶解选择性,N2的分子尺寸较小,通过膜孔的扩散比CH4快。
图9 展示了水相单体MPD 浓度对膜渗透选择性能的影响。在进料侧混合气总压力2~6 bar 范围内,相同压力下浓度为1.0%、1.5%和2.0%的膜N2和CH4渗透速率均随MPD 浓度升高而降低,这是由于MPD 浓度升高,膜厚度增加,导致气体扩散阻力增大。而MPD 浓度为2.5%的膜在2~3 bar 压力下N2和CH4渗透速率低于MPD浓度2.0%的膜,这也是由于MPD 浓度升高膜厚度增加,但随压力升高至超过4 bar,该膜的N2和CH4渗透速率急剧升高,分离因子急剧降低,这是由于TMC 浓度一定时,当MPD 浓度升高,反应区域内TMC浓度偏低,氨基浓度过量,形成许多带有端氨基的低分子量聚合物,反应生成的聚酰胺层交联度低,链段柔软、刚性较差且含有缺陷,气体分子更易透过。此外,图9也显示N2/CH4分离因子随MPD 浓度升高呈现先升高后降低的趋势,说明MPD浓度2.0%膜具有较致密且无缺陷的结构。
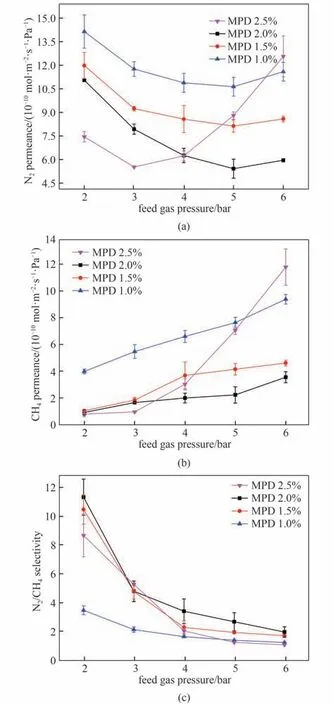
图9 MPD 浓度对膜渗透速率和分离因子的影响[油相中TMC的浓度为0.10%,进料气为N2/CH4混合气(体积比:50/50)]Fig.9 Effect of MPD concentration on membrane gas permselectivity[TMC concentration of 0.10%,feed gas:N2/CH4(50/50 by volume)]
图10 考察了MPD 浓度固定为2.0%时,TMC 浓度对膜渗透速率和选择性的影响。在相同测试压力下,随着TMC浓度升高,N2与CH4在膜中的渗透速率先降低后升高。这是由于水相溶液浓度一定,TMC 浓度较低时,反应区域内TMC 浓度偏低,界面聚合反应速率低,形成较疏松、孔径较大的聚合物膜,不利于筛分作用,随TMC 浓度升高后,反应速率加快,生成更加致密的分离层整体,导致了两种气体渗透速率降低;当TMC 浓度增至高于0.20%时,反应区域内TMC 浓度相对于MPD 浓度偏高,MPD分子很快会被反应掉,不能继续向有机相扩散,导致生成厚度较小且较疏松分离层,降低了膜的传质阻力,导致此后在压力一定时随TMC 的浓度升高,所制膜的气体渗透速率均升高。而如图10 所示可以观察到N2/CH4分离因子先升高后降低,分离因子在TMC 浓度为0.10%时达到最高。综上所述,当MPD 浓度为2.0%,TMC 浓度为0.10%时,酰氯和氨基配比最合适,活性分离层最为致密且无缺陷,故此时膜的选择性能最高。因此后续实验均采用该单体浓度,即MPD浓度为2.0%,TMC浓度为0.10%。
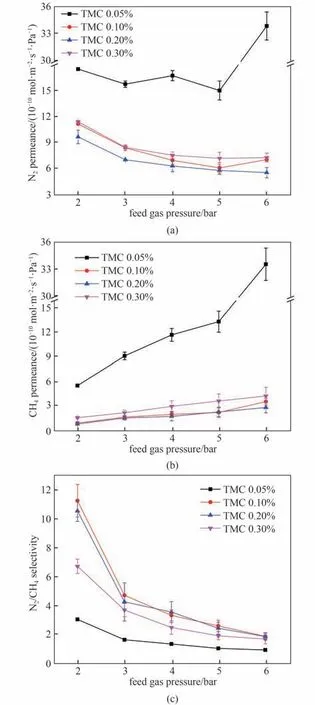
图10 TMC浓度对膜气体渗透速率和分离因子的影响[水相中MPD的浓度为2.0%,进料气为N2/CH4混合气(体积比:50/50)]Fig.10 Effects of TMC concentration on membrane gas permselectivity[MPD concentration of 2.0%,feed gas:N2/CH4(50/50 by volume)]
2.4 制膜条件对膜性能的影响
如图11 所示,相较初始膜,经过热处理的膜随热处理温度的升高,N2和CH4的渗透速率先降低后升高,在热处理温度为80℃时分离因子达到最大值。这是由于当热处理温度低于80℃时,热处理能够使初始膜中未反应的酰氯和氨基进一步反应,随着热处理温度升高促进反应程度,残留官能团继续交联,膜的交联度增加,减少了膜的缺陷,生成了更致密的聚酰胺分离层,更大程度阻碍了大分子气体的传输,提高了膜的筛分能力,从而使渗透速率降低,N2/CH4选择性提高。当热处理温度高于80℃,尽管较高的反应区温度增加了总反应速率,但过高的温度导致溶剂的挥发过快,膜收缩导致膜孔结构改变,无法使聚酰胺链段进一步交联,因此热处理温度升高,两种气体渗透速率也随之升高,N2/CH4选择性下降。
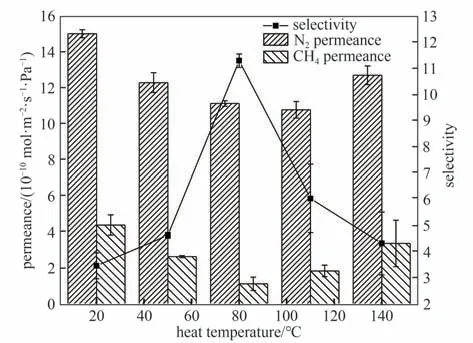
图11 热处理温度对膜气体传递性能的影响[水相中MPD的浓度为2.0%,油相中TMC的浓度为0.10%,进料气为N2/CH4混合气(体积比:50/50),测试条件:2 bar,22℃]Fig.11 Effects of heat treatment temperature on gas permselectivity of membranes[MPD concentration of 2.0%,TMC concentration of 0.10%,feed gas:N2/CH4(50/50 by volume),2 bar,22℃]
同样,如图12 所示,随着反应时间从0 增加到8 min,N2渗透速率约减少至初始值的45.5%,而CH4渗透速率约减少至初始值的19.6%。更长的反应时间可能导致初始膜中更多未反应的基团在反应区内进一步交联,形成额外的聚酰胺,并导致聚酰胺网络致密程度提高,封闭超薄膜中的缺陷,导致两种气体渗透速率均下降,但对较大气体分子的传输起到更大的阻碍作用,在反应4 min 时达到最大的N2/CH4分离因子。因此最终确定最佳制膜条件为在80℃下热处理4 min,后续实验均采用此条件。
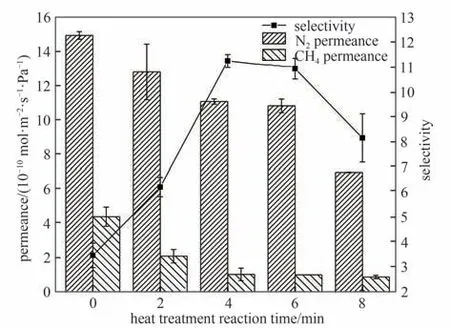
图12 热处理反应时间对膜气体传递性能的影响[水相中MPD的浓度为2.0%,油相中TMC的浓度为0.10%,进料气为N2/CH4混合气(体积比:50/50),测试条件:2 bar,22℃]Fig.12 Effects of heat treatment reaction time on gas permselectivity of membranes[MPD concentration of 2.0%,TMC concentration of 0.10%,feed gas:N2/CH4(50/50 by volume),2 bar,22℃]
2.5 MOF引入到油相或水相对膜性能的影响
界面聚合制备混合基质膜的过程中,选择将纳米填料引入到水相或油相中会产生不同的效果,进而会影响纳米填料在溶液中的分散并最终影响膜的性能。如图13 测试结果显示,在测试压力范围内,将纳米填料加入到水相中制膜相比加入到油相制 膜,N2/CH4分 离 因 子 更 高,N2与CH4渗 透 速 率 更低。推测这是由于颗粒加入水相后ZIF-90 中的醛基与MPD 分子或聚酰胺链端的氨基发生席夫碱反应,使其在溶液内更好分散,导致界面相容性增强;而将纳米颗粒加入油相后,油相溶液不与纳米颗粒发生反应,且向膜框内倒入油相溶液后,过量的油相溶液会迅速与膜表面的MPD 发生反应生成聚酰胺,不存在过量氨基去和纳米颗粒反应,因此成膜后易形成更多的缺陷。综上所述,将纳米颗粒加入到水相更有利于提升膜性能。故本文中后续的工作将采用纳米颗粒加入到水相的方法。
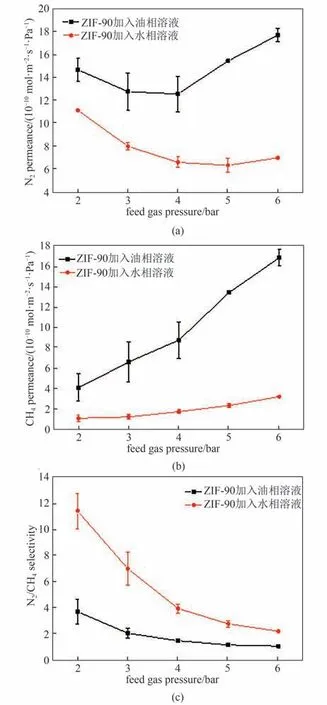
图13 MOF在油相或水相对膜的气体渗透选择性能的影响[水相中MPD的浓度为2.0%,油相中TMC的浓度为0.10%,颗粒添加量为0.10 g·L-1,进料气为N2/CH4混合气(体积比:50/50)]Fig.13 Effects of MOF in organic or aqueous phase on membrane gas permselectivity[MPD concentration of 2.0%,TMC concentration of 0.10%,nanoparticle content 0.10 g·L-1,feed gas:N2/CH4(50/50 by volume)]
2.6 MOF浓度对膜性能的影响
如图14 所示,对于任意的进料压力,随着ZIF-90 纳米颗粒添加量的增加,N2在膜中的渗透速率增大。例如在3 bar 的进料气压力下,随着纳米颗粒添 加 量 自0.10 g·L-1增 加 到0.65 g·L-1,膜 的N2渗透 速 率 由8.01×10-10mol·m-2·s-1·Pa-1上 升 至9.23×10-10mol·m-2·s-1·Pa-1。这 是 由 于 添 加 的 纳 米 颗 粒ZIF-90 孔径为0.35 nm[26],其引入增加了膜内小孔道的含量,由于CH4、N2分子的尺寸及形态差异,ZIF-90的孔道更有利于动力学直径较小的N2分子通过,因此引入颗粒可作为使N2快速渗透的传递通道,从而可以提升N2渗透速率。此外,与纯聚合物膜相比,颗粒的引入会干扰聚酰胺链的堆积,以致产生更多的由聚酰胺链堆叠形成的微孔,这些微孔也可为N2提供更多在膜内的传递通道,同时降低了N2传递阻力,提升了N2在膜中的渗透速率。

图14 ZIF-90/聚酰胺混合基质膜的渗透选择性能[水相中MPD的浓度为2.0%,油相中TMC的浓度为0.10%,进料气为N2/CH4混合气(体积比:50/50)]Fig.14 Gas permeation rate and separation factor of ZIF-90/polyamide MMMs[MPD concentration of 2.0%,TMC concentration of 0.10%,feed gas:N2/CH4(50/50 by volume)]
当测试压力为2~6 bar,ZIF-90纳米颗粒添加量小于0.50 g·L-1时,以及测试压力为2~4 bar,颗粒添加量为0.50 g·L-1时,混合基质膜CH4渗透速率均小于纯聚合物膜,N2/CH4分离因子均大于纯聚合物膜,这是因为纳米颗粒孔道尺寸小于CH4的动力学直径,CH4分子进入颗粒通道内的难度相比于N2更大,因此加入纳米颗粒后CH4渗透速率降低而N2/CH4分离因子提高;而当测试压力大于4 bar,颗粒添加量为0.50 g·L-1时,混合基质膜的CH4渗透速率大于纯聚合物膜,N2/CH4分离因子小于纯聚合物膜,这是由于CH4更容易与膜材料结合,且随着压力增大更多CH4进入膜内,聚酰胺链段间结合力和聚合物链段与纳米颗粒间结合力均减弱,导致了颗粒和聚合物之间产生了非选择性区域,相比于颗粒添加量较少的膜,更多CH4进入膜内会导致颗粒添加量较多的混合基质膜产生更多的非选择性区域,因此该膜在高压下CH4渗透速率显著增加。值得注意的是,由于颗粒的含量进一步增大,分离层中聚合物基质链段的堆积受到颗粒干扰,也导致了膜内出现非选择性缺陷,从而导致了CH4渗透速率的快速增加,分离因子迅速降低,分离性能减弱,例如当添加量0.65 g·L-1时,随着压力升高,CH4渗透速率迅速上升至2.81×10-9mol·m-2·s-1·Pa-1,N2渗透速率迅速上升至2.69×10-9mol·m-2·s-1·Pa-1,分离因子迅速降低到0.957,此时膜已不再优先渗透N2。此外,可以观察到当负载量小于等于0.30 g·L-1时,N2/CH4分离因子随添加量增加而增加,但当负载量超过0.30 g·L-1时,膜的N2/CH4分离因子随纳米颗粒添加量的增加而下降。此外,混合基质膜选择性随压力升高而下降,是因为所制备的混合基质膜中纳米颗粒添加量较少,渗透选择性能受界面聚合生成的聚酰胺本身性质的限制较为严重。在后续工作中会优化纳米材料和聚酰胺的相互作用,提高纳米材料添加量,从而突破聚酰胺本身性质对膜性能的局限,提高膜材料的耐压能力。在此基础上,还会针对N2的特点,进一步开发出具有更高N2选择性传输通道且与聚合物材料结合力良好的新型多孔材料,以制备高性能氮气优先渗透的N2/CH4分离膜。因此,综合考虑分离膜的渗透性和选择性,加入ZIF-90后膜的分离性能会有所提高,当纳米颗粒负载量为0.30 g·L-1时,膜具有最佳的气体分离性能,在进料气压力2 bar 下N2渗 透 速 率 为1.16×10-9mol·m-2·s-1·Pa-1,N2/CH4分离因子为16.6。
2.7 稳定性测试
采用N2/CH4混合气测试了ZIF-90 纳米颗粒含量为0.30 g·L-1的混合基质复合膜在进料气压力为3 bar 下性能随时间的稳定性,由图15 可以看出,该膜的渗透选择性能随着时间的延长基本不变,这表明,实验制备的混合基质复合膜性能具有良好的随时间稳定性。
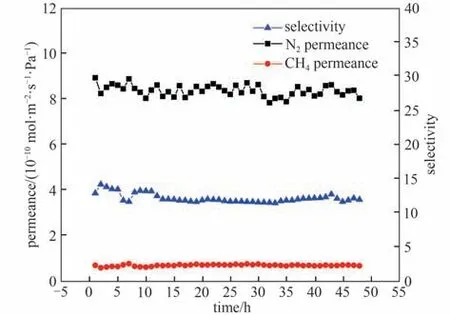
图15 膜的性能稳定性测试[纳米颗粒含量0.30 g·L-1,进料气为N2/CH4混合气(体积比:50/50),进料气压力:3 bar]Fig.15 Separation performance stability of the membrane[nanoparticle content 0.30 g·L-1,feed gas:N2/CH4(50/50 by volume),feed gas pressure:3 bar]
2.8 与文献中其他膜的性能比较
表2 将本文实验制备的膜与文献中N2/CH4分离膜的渗透选择性能进行比较。由表可以看出,ZIF-90 负载量为0.30 g·L-1的聚酰胺混合基质复合膜与文献中报道的其他氮气优先渗透的N2/CH4分离膜相比,具有更高的N2/CH4分离选择性。
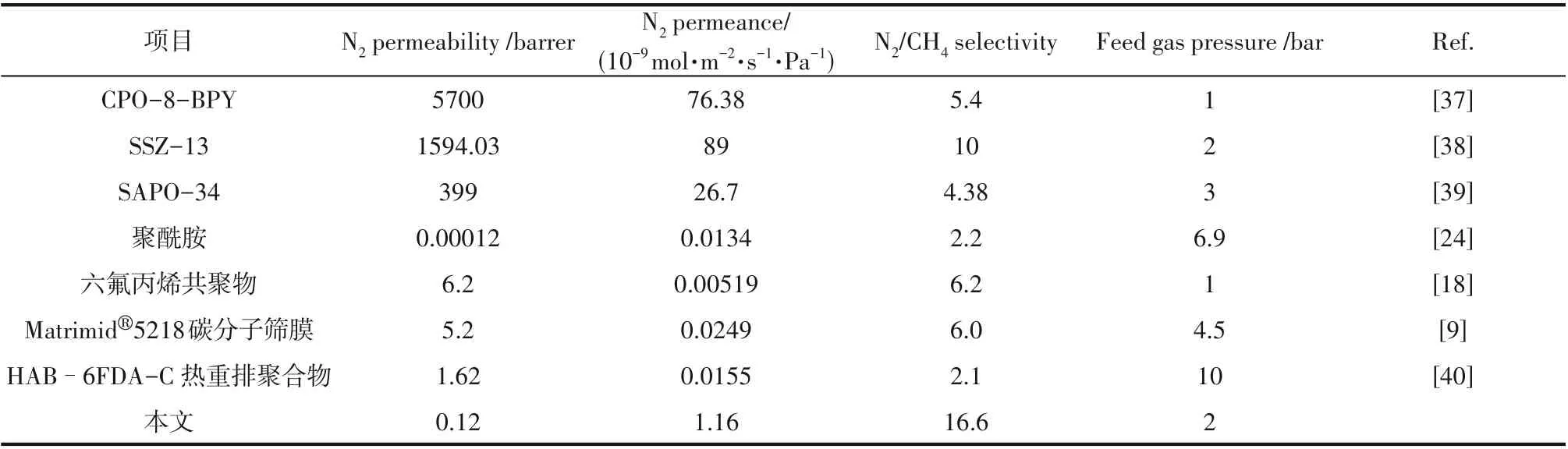
表2 与文献中其他膜的N2/CH4分离性能的比较Table 2 N2/CH4 separation performance comparison with other membranes in references
3 结 论
以MPD 为水相单体,TMC 为油相单体,采用界面聚合法成功制备出较致密的聚酰胺复合膜,通过引入孔径可以允许N2分子通过而不允许CH4分子通过的纳米颗粒ZIF-90,制备成混合基质复合膜。探究了单体浓度以及颗粒含量对膜分离性能的影响,并通过优化这些制膜条件制备出分离性能较优的复合膜。在所制成的混合基质膜中,当水相溶液中MPD 浓度为2.0%并添加0.30 g·L-1纳米颗粒ZIF-90,油相溶液中TMC含量为0.10%时,制备的膜致密无缺陷且膜内含有固定的N2传递通道,因而N2/CH4分离因子最高,N2渗透速率也较高,即在进料压力2 bar时,膜的N2渗透速率为1.16×10-9mol·m-2·s-1·Pa-1,N2/CH4分离因子为16.6。
猜你喜欢
广州化工(2022年19期)2022-11-09
材料科学与工程学报(2022年1期)2022-02-28
纺织学报(2021年4期)2021-12-06
农业研究与应用(2021年3期)2021-08-23
纺织导报(2021年4期)2021-05-06
煤矿爆破(2021年4期)2021-03-11
煤矿爆破(2020年3期)2020-12-08
燃料化学学报(2020年3期)2020-05-07
安徽农业科学(2017年1期)2017-07-10
火炸药学报(2017年3期)2017-06-28
