
TGF-β参与黄斑前膜形成的研究进展
2022-08-09 07:28冯家桢邢怡桥
国际眼科杂志 2022年8期
冯家桢,贺 涛,邢怡桥
0引言
黄斑前膜(epiretinal membrane, ERM)是一种以发生于视网膜内表面的纤维细胞膜为特征的视网膜疾病。ERM可分为不伴明显原发疾病的特发性黄斑前膜(idiopathic epiretinal membrane, iERM),及继发于眼外伤、眼内炎、视网膜血管疾病、或眼部手术等的继发性ERM[1]。多数ERM是无症状且长期无进展的,少数ERM的整体病程是逐渐进展的[2]。ERM的诊断依赖于裂隙灯下所见眼底及OCT检查[3-4]。ERM在病理上表现为视网膜内表面上形成的病理性纤维细胞膜[1],绝大多数ERM主要由两部分组成,即细胞与细胞外基质(extracellular matrix, ECM)[5-6]。在ERM的早期病理表现中可见神经胶质细胞明显增多,但此时期患者多无明显症状,仅在裂隙灯检查下可见黄斑玻璃纸样反射。而在ERM的晚期病理表现中肌成纤维细胞明显增多,膜表现收缩特性,OCT下表现为视网膜前黄斑纤维化,此时期患者多有严重视力下降及视物变形症[7-8]。早期ERM通常无需随访。而晚期ERM患者由于持续性的视力下降及严重视物变形常需行玻璃体切割术剥离ERM[9]。
1ERM
目前对ERM的研究多集中于病理及细胞水平。就iERM而言,随年龄增长逐渐进展的玻璃体液化所导致的玻璃体后脱离(posterior vitreous detachment, PVD)可能是其发生的始动因素[10],这一观点已得到多数学者的认可。PVD可导致内界膜(internal limiting membrane, ILM)损伤,并在其表面形成微渗漏孔,这使得视网膜色素上皮层(retinal pigment epithelium, RPE)细胞、Müller细胞、小胶质细胞等细胞得以向ILM表面迁移。其次,PVD所导致的玻璃体劈裂也使得玻璃体细胞可以迁移至ILM表面[11]。在早期iERM的病理表现中,神经胶质细胞占据主导地位,其中以来源于星形胶质细胞、Müller细胞等细胞的转分化的细胞为主[12]。但在晚期iERM的病理表现中肌成纤维细胞占主导地位[13],目前认为晚期iERM中的肌成纤维细胞来源于Müller细胞、玻璃体细胞、RPE细胞的转分化、肌成纤维细胞分泌收缩蛋白并沉积胶原蛋白,使晚期iERM呈现收缩牵拉特性[14]。继发性ERM同样可能与PVD有很大关联,但其与原发病的联系更为紧密,已有的研究表明继发性ERM本质上是一种愈合反应,这种反应可以经由缺氧,感染等病因通过激活转化生长因子-β(transforming growth factor-β, TGF-β)相关通路促进细胞增殖及细胞间质的合成从而导致ERM的形成[15]。还有研究表明IL-6、IL-8、单核细胞趋化蛋白-1等炎性细胞因子的上调及巨噬细胞、T细胞、B细胞等免疫细胞的活化参与了部分继发性ERM的发生发展[16]。
2TGF-β
TGF-β是高度多效细胞因子,其在哺乳动物中存在三种同工型(TGF-β1、TGF-β2和TGF-β3),它们包含高度保守的区域,仅在几个氨基酸区域中存在差异,且都通过相同的受体信号通路在伤口愈合、血管生成、免疫调节、癌症及纤维化疾病中扮演重要角色[17]。TGF-β同源二聚体与潜伏相关肽(latency associated peptide,LAP)相互作用,形成小潜伏复合体(small latent complexes,SLC),随后与TGF-β结合蛋白(latent transforming growth factor β binding proteins,LTBP)结合,形成大型潜在复合体(large latent complex,LLC)。LLC以非活性形式分泌到细胞外基质中,阻止TGF-β与其受体结合。LLC从基质中释放后,LAP与整合素αvβ6等结合后释放活性TGF-β[18]。TGF-β有三种存在于胞膜的受体(TGFβRⅠ、TGFβRⅡ和TGFβRⅢ)。TGFβRⅠ和TGFβRⅡ的细胞质结构域中都含有丝氨酸/苏氨酸蛋白激酶,而TGFβRⅢ没有激酶活性。TGF-β与TGFβRⅡ的结合以及与TGFβRⅠ的异源四聚化通过下游Smad启动细胞内信号传导。TGFβRⅢ作为辅助受体发挥作用,以增加配体与TGFβRⅡ的结合效率[19]。活化的TGF-β经受体转运进入胞质后激活Smad信号通路,介导Smad2/3的磷酸化,磷酸化后的Smad2/3与Smad4结合后进入细胞核内调控相关基因表达[20]。除上述经典TGF-β-Snail途径外,TGF-β下游还存在着Wnt/β-catenin通路、Snail通路等其他信号途径,调控多种生理病理过程[21]。
3TGF-β与ERM
现阶段学术界多将ERM视为始动于PVD的视网膜内界膜层面的损伤愈合反应或纤维化疾病。近年来对ERM成分的多项免疫组化研究也表明了TGF-β及其相关通路在ERM发病中的重要地位。在TGF-β所参与的ERM的发病机制中,可分为非经典的TGF-β-Snail途径与经典的TGF-β-Smad途径。
3.1TGF-β-Snail途径与上皮-间质转化上皮-间质转化(epithelial to mesenchymal transition, EMT)是1960年代由Elizabeth Hay首次提出的概念,指上皮细胞在胚胎发育时期及病理状态下基因表达谱及表型均发生改变,转化为具有间充质特点的具有迁移能力的间叶样细胞的过程[22-23]。EMT使细胞黏附分子的表达下调,角蛋白细胞骨架转变为波形蛋白,从而使上皮细胞失去极性与上皮表型,获得较高的迁移与侵袭、抗凋亡和降解细胞外基质等特性。EMT在胚胎发育(1型EMT),组织修复及纤维化(2型EMT)及上皮性肿瘤的转移与侵袭(3型EMT)方面发挥重要作用[24]。而继发性ERM本质上是一种损伤愈合及纤维化反应,2型EMT在其发生及发展中起着重要作用。
研究表明Snail、Slug、ZEB1、SIP1和Twist等转录因子参与了EMT的调控,其中以TGF-β-Snail途径的作用最为关键[25-26]。TGF-β介导Snail与E-钙黏着蛋白基因(E-cadherin)启动子中的特定DNA序列E-boxs结合并抑制E-cadherin的转录,下调细胞黏附蛋白E-钙黏着蛋白表达,而E-钙黏着蛋白的下调被视为EMT的标志[27]。
一般认为在继发性ERM中EMT的发生发展过程是由源于视网膜脱离(retinal detachment, RD)后经历增生性玻璃体视网膜病变(proliferative vitreoretinopathy, PVR)的RPE细胞[28],RD后由于视网膜的断裂与血-视网膜屏障的破坏导致RPE细胞直接暴露于TGF-β,导致细胞内Snail合成增多,作用增强进而导致EMT,使得RPE细胞转变为成纤维细胞,分泌ECM,从而形成ERM[29]。Li等[30]的一项研究发现在沉默Snail后间充质标志物——纤连蛋白及a-肌动蛋白(alpha-smooth muscle Actin,a-SMA)减少,上皮标志物——E-cadherin及ZO-1增加,这进一步证明在继发于PVR的ERM中Snail是RPE细胞在TGF-β介导下发生EMT的重要调控因子。
在晚期iERM中,其特征细胞成分肌成纤维细胞与2型EMT关系密切[31-32],但iERM中转化为肌成纤维细胞的Müller细胞却并非上皮来源[33-34],Kanda等[35]研究了Müller细胞-间质转化(GMT)作为EMT替代机制的可行性。通过实验,Atsuhiro Kanda发现与BMP-4、CTGF等细胞因子相比,TGF-β1刺激下2型EMT标记物(α-SMA,SM22,Ⅰ型胶原)上调明显,故将TGF-β1作为潜在的2型EMT诱导剂。进一步测定TGF-β1刺激后EMT相关标记物水平,发现Snail表达明显上调,并且敲低Snail mRNA耗竭Snail抑制了TGF-β1诱导的EMT相关基因转录的上调。后续实验进一步揭示了Snail在Müller细胞间质转化中赋予其迁移能力及丧失胶质表型的作用。综上所述,Müller细胞可经历由TGF-β-Snail轴驱动的2型EMT,并提出了Müller细胞GMT这一新概念,而其他可发生ERM的非上皮源性细胞如玻璃体细胞及其他胶质细胞是否也有类似的转化过程还需进一步研究。
3.2TGF-β-Smad途径RPE细胞除经TGF-β-Snail轴外还受到很多其他细胞因子及通路如p38-MAPK途径、PKA、HSP90和MDM2等的调控经历2型EMT,而这些因子都通过对经典的TGF-β-Smad途径的影响进而促进RPE细胞迁移至ILM表面形成ERM。
3.2.1p38-MAPK 丝裂原活化蛋白激酶(mitogen activated protein kinase, MAPK)由一组级联活化的丝氨酸-苏氨酸蛋白激酶蛋白组成,通过依级联反应将上游信号传递至下游应答分子。p38是MAPK家族的重要成员,参与炎症的调控及细胞增殖、分化、凋亡等过程[36-37]。
在PVR导致的ERM中,经历EMT的活化RPE细胞可达95%[38-39],已有研究表明多种细胞因子在诱导RPE细胞活化中起作用,其中以TGF-β和TNF-α的研究最为成熟[40-41],Shirasawa等[42]发现TNF-α可通过p38-MAPK途径损伤RPE屏障功能进而导致其经历EMT,而在肺泡上皮细胞与小肠上皮细胞的EMT中TGF-β与TNF-α有协同作用[43]。Schiff等[44]通过对进行TGF-β和TNF-α共同处理(TNT)所形成的表现特殊收缩特性的体外成人RPE(adult human RPE,ahRPE)细胞聚集体进行研究,对比对照组、敲除p38基因组及使用p38抑制剂SB202190组收缩性物质的含量,结果表明敲除p38基因组及使用p38抑制剂SB202190组收缩性物质的含量下调甚至逆转,这证明了p38-MAPK途径是TGF-β和TNF-α下游信号通路的主要信号传导节点,并且他们将采集到的患者ERM样品进行转录组学分析,结果也呈现p38-MAPK途径的富集。2018年Chen等[45]发现Plumbagin(PLB)可通过下调p38-MAPK信号通路相关分子表达来抑制PVR中RPE的增殖特性,展现了抑制p38-MAPK途径在ERM临床治疗方面的可观前景。
3.2.2TGF-β与HSP90 在iERM中,由于ECM中各型胶原及纤连蛋白等基质成分的存在,使其具有纤维化疾病的特点[46-47],而TGF-β介导了纤维化疾病的进展,免疫组化表明iERM中TGF-β1及其受体的过表达[48-49]。在TGF-β通路中,TGF-β与2型TGF-β受体结合后激活1型TGF-β受体[50-51],进而激活下游Smad途径,Smad是TGF-β通路的重要细胞质介质,Smad2/3磷酸化后与Smad4结合并转移至核内,调节多种基因表达,最终调控αSMA和Ⅰ型胶原等促进纤维化的蛋白的表达[52-53]。上述通路中存在包括热休克蛋白90(HSP90)在内的多种调节剂。已有报道证明HSP90在其他纤维化疾病中的作用,即HSP90能够使2型TGF-β受体免受降解进而维持其磷酸化后的活性,从而促进纤维化物质的表达[54]。Sethi等[55]研究了iERM中HSP90在TGF-β通路中的调控机制。结果显示在表达2型TGF-β受体的iERM细胞中发现较高表达的HSP90,同时观察到的Smad2/3、Smad4表达上调,表明Smad途径的激活得到了增强。目前对于ERM中HSP90的更多机制还需进一步研究,但可将其视为潜在的治疗靶标。
3.2.3TGF-β与蛋白激酶A 蛋白激酶A(cAMP-dependent protein kinasea,PKA)是一类依赖于cAMP的蛋白激酶,激活后可导致多种蛋白质磷酸化,在细胞生长、分化、凋亡及ECM分泌等过程中发挥重要作用[56-57]。RPE细胞的EMT已被认为是PVR后ERM形成的主要原因,其中TGF-β是EMT的一种重要调节剂[58-59]。TGF-β诱导的EMT受到多种信号通路及分子的串扰调节,PKA是串扰的重要组成部分。早先的研究已经证实在肾小球系膜细胞及胰腺腺泡细胞等细胞中PKA能够调节TGF-β诱导的EMT[60-61],Lyu等[62]研究了PKA在PVR后RPE细胞EMT中的作用,实验中通过对PVR患者ERM的PKA C亚基(PRKACa)表达的检测发现PRKACa在ERM样品中表达量明显上升,且与EMT标记物α-SMA和上皮标记物CK8强烈共定位,结果表明PKA在经历EMT的RPE细胞中被激活。随后,实验者向PVR模型大鼠玻璃体腔内注射选择性PKA抑制剂H89,可观察到PVR进展的延缓及眼底结构的改善,并且视网膜电图(electroretinogram, ERG)b波波幅也得到了改善这表明H89对大鼠的视觉功能有一定的保护作用。在后续实验中,Lyu等[62]又进一步验证了H89对TGF-β途径中有重要作用的Smad2/3没有影响,不会引起其磷酸化或核异位,但阻断了TGF-β1对抑制性信号Smad6的下调作用,说明H89增强了Smad6的抑制作用。这一结果揭示了PKA抑制剂在治疗ERM方面的潜在可能。
3.2.4TGF-β与小鼠双微体2 小鼠双微体2(mouse double minute 2,MDM2)是一种致癌基因所编码的蛋白,它可以作为P53-E3-泛素的连接酶促进抑癌基因编码的P53蛋白通过泛素途径降解,MDM2在p53依赖或非依赖途径的肿瘤EMT中占有重要地位[63-64]。MDM2基因中有两个启动子,启动子1(P1)是管家启动子,而位于内含子1的启动子2(P2)则可被Smad或特异性蛋白1(Sp1)激活,进而响应包括TGF-β在内的各种刺激调控[65-66]。Sp1与P2的亲和力增加可上调MDM2的表达,进而诱导RPE的EMT进程,最终导致罹患包括ERM在内的PVR的风险增高[67]。因此,抑制P2驱动的MDM2表达在预防ERM的发生发展中有着巨大的潜力。成簇的规则间隔短回文序列相关蛋白9(Cas9)是一种特异性极高的遗传编辑工具,可通过阻断转录延伸、阻止RNA聚合酶或转录因子与真核生物DNA结合靶向干扰真核生物基因组[68-71]。Liu等[72]用含有两个突变的靶向MDM2 P2 dCas9(dCas9)的慢病毒转染已受TGF-β2诱导而发生EMT的人RPE(ARPE-19),发现dCas9阻断了TGF-β2诱导的MDM2表达进一步的研究发现,dCas9也可抑制MDM2在非刺激条件下的基础表达,暗示了高选择性的dCas9疗法在治疗继发于PVR的ERM方面的潜在价值[73-74]。
4总结与展望
TGF-β自1981年在转化大鼠肾成纤维细胞的培养物中被发现后,对其功能及上下游信号通路的研究一直是医学生物领域的热点。经过多年的研究,TGF-β在纤维化、肿瘤、炎症、免疫调节等生理病理过程中的作用机制被逐一揭示。而ERM作为PVD后视网膜各细胞经历EMT而形成的纤维细胞膜,TGF-β通路及其相关串扰因子在其中扮演重要角色(图1)。
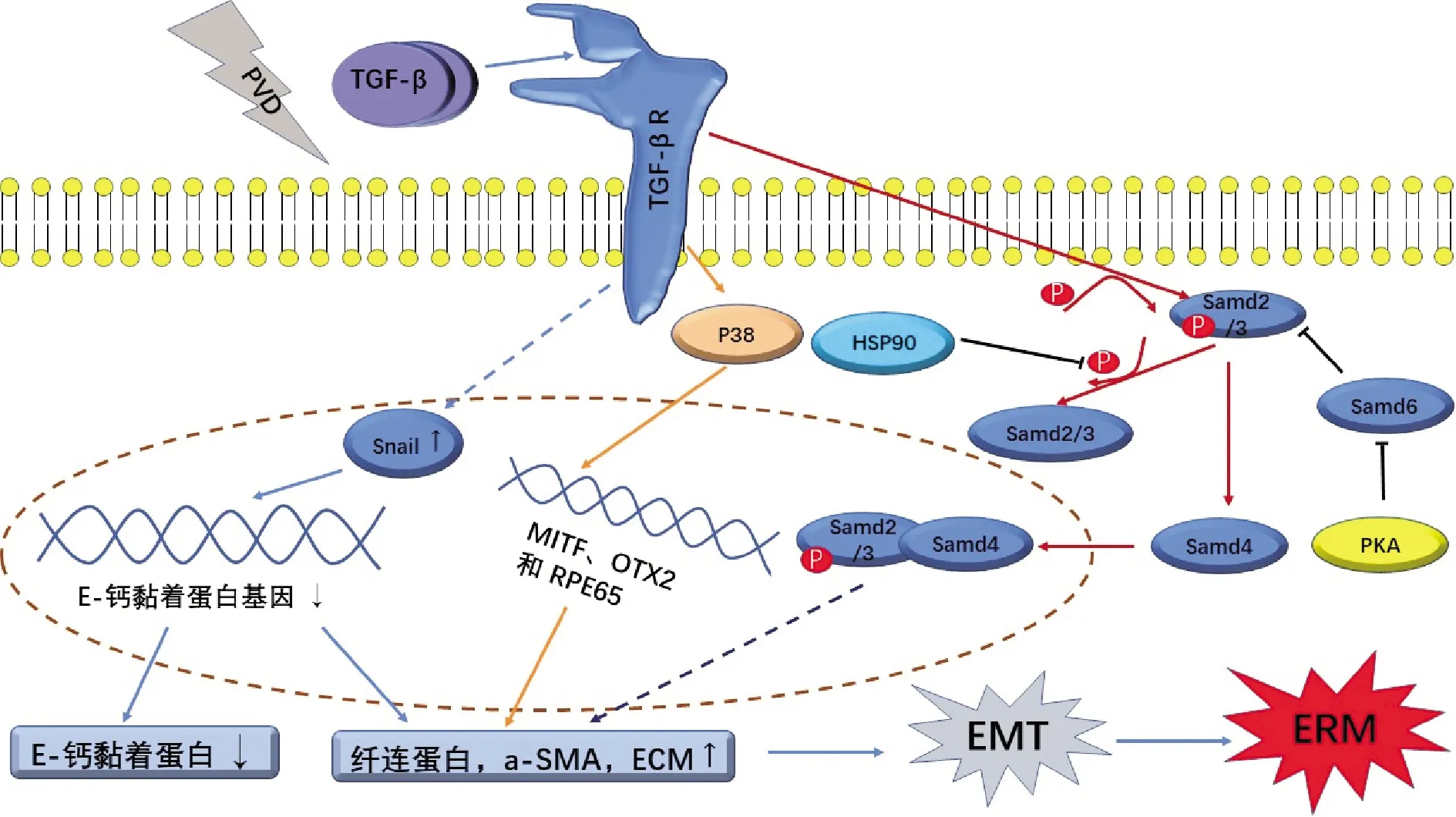
图1 TGF-β通路参与ERM形成的示意图 TGF-β诱导Snail合成上调,下调E-钙黏着基因表达,介导EMT。TGF-β激活P38通路,增强MITF、OTX2和RPE65等基因效应,上调纤连蛋白、α-SMA、ECM。活化的TGF-β经受体转运进入胞质后激活Smad信号通路,介导Smad2/3的磷酸化,磷酸化Smad2/3与Smad4结合后进入细胞核内调控相关基因表达。HSP90抑制Smad2/3去磷酸化,增强Samd2/3途径效应。PKA通过阻断Smad6对Smad2/3的抑制作用增强经典Samd通路效应。
当然,ERM的形成不仅依靠TGF-β的调节,越来越多的分子及通路被发现:如在增殖期糖尿病视网膜病变(proliferative diabetic retinopathy,PDR)患者的ERM中,肿瘤坏死因子超家族成员15(TNFSF15)与成纤维细胞生长因子诱导分子14(FN14)结合后可在多种细胞(血管内皮细胞,单核细胞、巨噬细胞和成肌纤维细胞)内激活核因子NF-κB信号转导途径[75],上调包括基质金属蛋白酶9(matrix metalloproteinase-9,MMP-9)、细胞间黏附分子1(intercellular adhesion molecule-1,ICAM-1)、E-选择素、IL-6、IL-8等多种促炎因子的表达,促进炎症反应并诱导胶质细胞增生,形成纤维细胞膜,值得一提的是,PDR中以NF-κB为核心的炎症信号通路上Wip1等相关分子在不断地被发现完善[76],而NF-κB在介导PDR患者继发性ERM形成过程中是否与TGF-β通路有串扰有待进一步的研究证实;Zhang等[76]研究中发现,ROCK信号通路在DR缺氧和氧化应激诱导的Müller细胞损伤中起着关键作用,ROCK途径的激活可以导致Müller细胞形态改变,增加迁移率并刺激细胞活性氧(reactive oxygen species,ROS)的生成[77-78],进而促进Müller细胞在ILM上形成ERM。
如今ERM的发病率逐渐提高,已经成为老年人视力损害的一大病因[79]。虽然玻璃体切除术合并ILM剥离这一一线治疗方案已成为有症状ERM的首选治疗,但手术具体入路选择的争议,染料的安全性不高,术后并发症较多及ERM复发风险较高等问题仍有待解决[80-82]。现阶段学术界尚未就ERM的发病的分子机制达成一致,对其分子机制的进一步研究不仅能够增进我们对ERM的理解,发现发病机制中重要的分子靶点也能为临床选择新治疗手段提供思路。众所周知,抗VEGF治疗自2004年进入眼科范畴以来,通过对相关疾病分子机制的不断完善,新型药物层出不穷,已成为玻璃体切除术外治疗DR等疾病的重要手段,且相较玻璃体切除术,玻璃体腔注射给患者带来的不适也大大减少[83]。而TGF-β在两型ERM的发生发展中虽占据重要地位,但由于TGF-β作为一种多功能的分子,单纯抗TGF-β治疗虽然能抑制ERM,但同样阻碍了细胞其他生理活动。因此,进一步对整个TGF-β通路相关分子的完善,精准抑制特异性高的分子,如Yoshida等[84]发现通过Smad4的去乙酰化诱导RPE细胞下调α-SMA并抑制TGF-β2诱导EMT的白藜芦醇等新靶点药物的将为ERM非玻璃体切除治疗及预防带来巨大的变革。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国现代医生(2022年21期)2022-08-22
皮肤性病诊疗学杂志(2022年3期)2022-08-01
传染病信息(2022年3期)2022-07-15
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2021年8期)2021-08-13
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
分析化学(2017年12期)2017-12-25
