
吲哚类抗癌药物的研究进展
2022-08-09 05:46:56汤晟孙鑫陈铮乔丹江西科技师范大学江西省重点分子实验室南昌330013
中南药学 2022年1期
汤晟,孙鑫,陈铮,乔丹(江西科技师范大学 江西省重点分子实验室,南昌 330013)
恶性肿瘤严重影响着人们的生命健康,据世界卫生组织统计,全世界每年因恶性肿瘤死亡的人数约占总死亡人数的25%[1-2]。癌症的发病率一直处于上升趋势,尽管现在有许多针对癌症的药物和治疗手段,但其依然是人类健康的最大威胁之一。
吲哚或1H-苯并[b]吡咯是一种有机化合物,分子式为C8H7N,其由六元苯环和五元含氮吡咯环组成,是一种富含π键的杂芳烃。吲哚骨架广泛存在于自然界的动物、植物和微生物激素中[3]。研究发现,许多吲哚类化合物都具有抗菌、抗炎、抗组胺、抗氧化、抗糖尿病、抗病毒、抗胆碱酯酶和抗癌等药理作用[4]。天然吲哚类药物主要有褪黑素(1,melatonine,MT),属于吲哚杂环类化合物,其化学名是N-乙酰基-5-甲氧基色胺,是一种重要的生理性肿瘤抑制剂[5-6],广泛存在于动植物体内,具有很好的抑制乳腺癌、前列腺癌、结肠癌等多种肿瘤的功能[7]。靛玉红(2)是一种吲哚的缩合产物,该化合物对慢性粒细胞白血病具有明显的抑制作用,且具有临床疗效可靠,毒副作用小,对骨髓无明显抑制作用等特点[8]。长春新碱(3)是catharanthine 环和vindoline 环以碳桥相连的二聚吲哚结构,高浓度长春新碱类化合物能诱导微管解聚,进而让癌细胞死亡[9]。长春新碱已被广泛用于治疗癌症,包括霍奇金氏病、非霍奇金淋巴瘤、卡波西肉瘤、乳腺癌和睾丸癌[10]。海洋类生物碱eudistomin(4)对p-388 淋巴瘤细胞具有抗增殖活性,有望作为抗癌药物设计的先导化合物。此外,双吲哚生物碱dragmacidin(5)、hyrtinadine(6)、nortopsentin(7)、topsentin(8)和hyrtiosins(9)都具有一定的细胞毒性[11](见图1)。
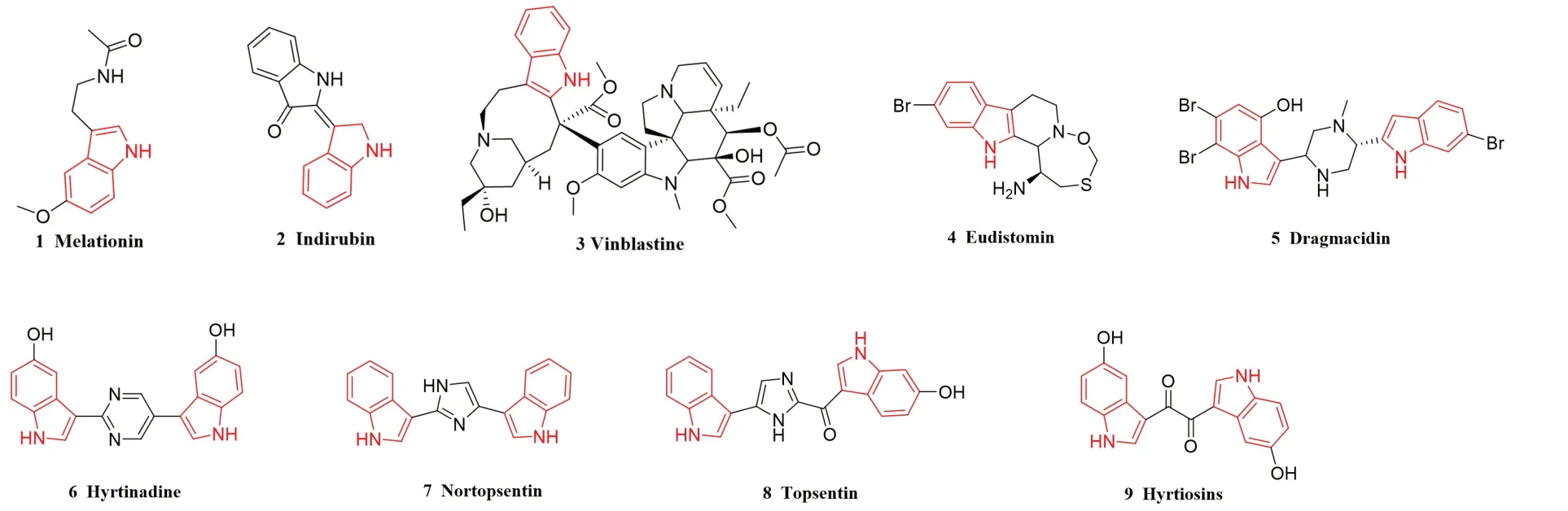
图1 含有吲哚结构的具有天然抗癌和细胞毒性的化合物Fig 1 Natural anticancer and cytotoxic compounds with indole structures
目前,已经上市的吲哚类抗癌药物有舒尼替尼(10,sunitinib)、阿来替尼(11,alectinib)、帕比司他(12,panobinostat)和奥西替尼(13,osimertinib)(见图2)。舒尼替尼是血小板内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)和血小板衍生生长因子受体(platelet-derived growth factor receptors,PDGFR)的多靶点受体酪氨酸激酶抑制剂,用于治疗胃肠间质瘤及晚期肾细胞癌[12];奥西替尼和阿来替尼分别是用于治疗非小细胞肺癌(non-small cell lung cancer,NSCLC)的表皮生长因子受体(epidermal growth factor receptor,EGFR) 与间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)抑制剂;帕比司他则为治疗多发性骨髓瘤的组蛋白去乙酰化酶(histone deacetylase,HDAC)抑制剂药物。近年来,研究者们针对不同靶点又设计合成了诸多吲哚类抗癌药物,本综述概括了近5年来吲哚类抗癌药物的研究进展。
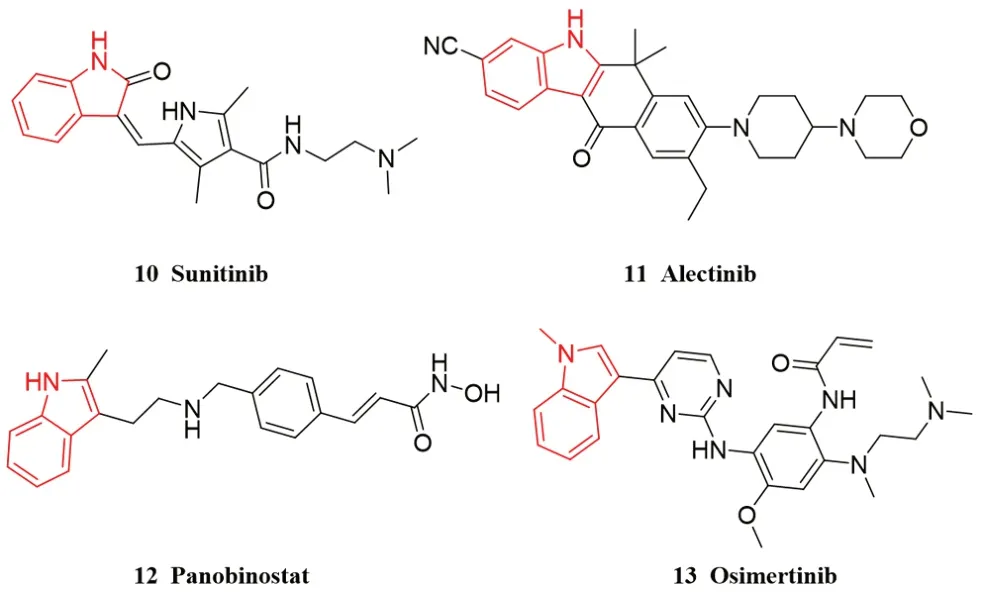
图2 已上市的吲哚类抗癌药物Fig 2 Indole anticancer drugs on the market
1 吲哚类抗癌药物
1.1 吲哚类EGFR 抑制剂
肺癌的病死率约占世界癌症病死率的33.3%,NSCLC 约占肺癌病例的85%。EGFR 是一种糖蛋白,属于受体型酪氨酸激酶家族的一类,用于控制细胞的增长,正常情况下EGFR 的作用时间短且受到严密的控制,它在行使完功能后,就会被关闭。若EGFR基因突变,会导致EGFR 不能正常关闭,无休止地刺激肿瘤细胞生长,最终导致肿瘤的发生。研究表明,NSCLC 的肿瘤中存在EGFR 的高度表达[13]。目前,用于治疗肺癌的酪氨酸激酶抑制剂类药物有贝伐单抗、西妥昔单抗、厄洛替尼、吉非替尼、奥西替尼和阿法替尼,虽然在初始治疗期间临床效果不错,但耐药性问题仍然无法解决[14]。
含有单吲哚结构的奥西替尼(13,osimertinib,AZD9291)作为第三代EGFR 抑制剂,主要针对具有EGFRT790M突变的患者。奥西替尼选择性高,对含T790M/L858R 突变和外显子19 缺失的EGFR 抑制活性强,其IC50值分别是11.4 nmol·L-1和12.9 nmol·L-1,而对野生型EGFR 的IC50值为494 nmol·L-1[15]。奥西替尼是不可逆的EGFR 抑制剂,能与分子靶点形成共价键,所以理论上应答持续更久,产生耐药的机会相应减少。但是,最近有报道称,奥西替尼也出现了C797S 突变耐药问题。C797位于EGFR基因第20 号外显子编码的酪氨酸激酶结合域,是EGFR 蛋白与抑制剂结合的关键靶点[16](见图3),该位点的突变导致三磷酸腺苷(ATP)竞争性靶向抑制剂无法阻止EGFR 蛋白与ATP 结合,进而无法抑制下游信号通路的持续异常活化。
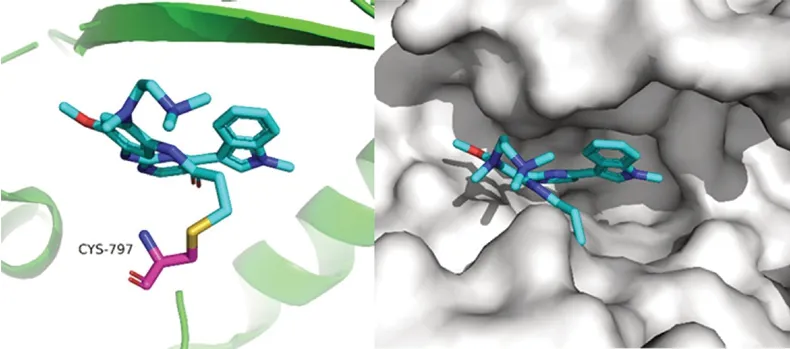
图3 奥西替尼与EGFR 蛋白(PDB:6JWL)的共晶结构模拟Fig 3 Co-crystal structure simulation and surface view of osimertinib with EGFR(PDB:6JWL)
Showalter 等[17]研究发现,N-(3-溴苯基)-6-甲氧基-9H-嘧啶基-[4,5-b]吲哚-4-胺表现出优异的抗肿瘤活性,可能是有效的EGFR 抑制剂。Zhang 等[18]在此基础上,将嘧啶并[4,5-b]吲哚处进行简化开环并引入具有活性的甲酰胺基团,引入已被证实具有潜在EGFR 抑制活性的苄氨基团和抗肿瘤活性基团2-呋喃甲氨基,设计合成了一系列N-(呋喃-2-甲基)-1H-吲哚-3-甲酰胺衍生物(见图4)。以厄洛替尼(erlotinib)为阳性对照,用MTT 法测定了目标化合物的体外抗增殖活性,结果发现在吲哚5 位C 原子处引入庞大芳基的化合物14 抑制效果显著(人非小细胞肺癌细胞A549:IC50=5.61 μmol·L-1,人宫颈癌细胞HeLa:IC50=5.33 μmol·L-1,人结肠癌细胞 SW480:IC50=10.48 μmol·L-1)。共晶结构模拟图揭示了化合物14 与EGFR(PDB:1M17)的连接方式, 化合物14 与Gly697、Gln767、Leu768、Thr830 和Asp831 形成氢键,与残基Lys721、Thr766、Leu694、Val702 和Ala719 形成较弱的疏水相互作用(见图5),苄氨基的引入恰好位于关键活性位点。因此,化合物14 是潜在的EGFR 抑制剂,具有一定的应用前景。
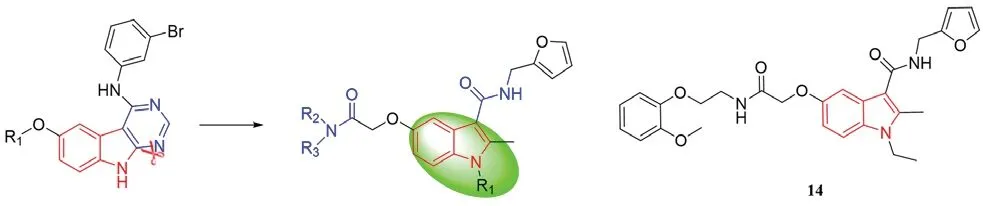
图4 化合物14 的合成Fig 4 Synthesis of compound 14
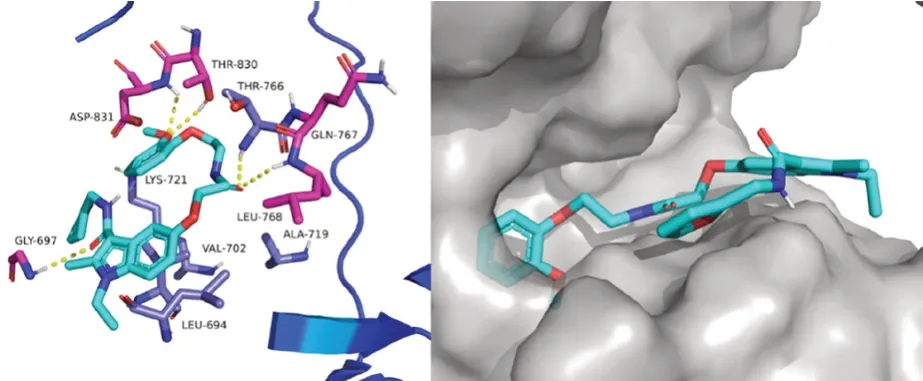
图5 化合物14 与EGFR 的共晶结构模拟(PDB:1M17)Fig 5 Co-crystal structure simulation and surface view of compound 14 with EGFR(PDB:1M17)
吲哚-3-甲醇(indole-3-carbinol,I3C,见图6)类化合物在十字花科蔬菜(西蓝花、球芽甘蓝、卷心菜等)中含量相对较高,I3C 消化后的产物是一种双吲哚衍生物。研究发现,该结构具有抗癌、抗氧化和抗动脉粥样硬化等作用[19-21]。在双吲哚衍生物的基础上又设计合成了一系列二吲哚-2-二硫化物,可以选择性地抑制EGFR 来阻断肿瘤细胞的信号转导,从而抑制癌症的发展[22]。
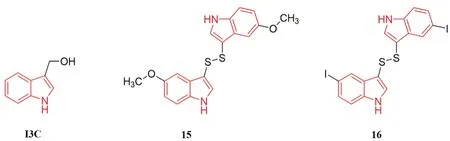
图6 吲哚类EGFR 抑制剂Fig 6 EGFR inhibitors with indole structure
Żołek 等[23]通过计算机模拟,对Maciejewskaa等[24]设计的S-S 取代亚甲基的双吲哚衍生物进行了毒性测试(见表1)。结果显示,所有化合物毒性剂量均低于推荐最大治疗剂量值3.16 mg/(kg·d)。hERG 钾离子通道抑制的可能性(hERG_filter)和达到50%抑制效果时抑制剂浓度的对数值(pIC50)显示,化合物15、16 无心脏毒性,具有一定的研究价值。在肝脏毒性测试中,除了有甲氧基取代的化合物15,其余化合物均由于谷氨酰转肽酶(GGT)、碱性磷酸酶(Alk-Phos)、乳酸脱氢酶(LDH)、血清谷草转氨酶(SGOT)和谷丙转氨酶(SGPT)水平的增加而被评估为对肝脏功能有毒性;计算机模拟自由焓计算与人原髓细胞白血病细胞HL-60、人前列腺癌细胞DU-145 的细胞毒性实验显示,带有I 原子的化合物16 与EGFR 的作用非常强,IC50值为0.87 μmol·L-1(见表2)。化合物15 无肝毒性,但与EGFR 的结合能力很小,所以成为EGFR 抑制剂的潜力有限。综上,双吲哚衍生物具有一定活性的同时也具有一定程度的毒副作用;在结构改造方面,吲哚部分C-5 位置的I 原子取代后对EGFR 激酶的抑制显著提升。
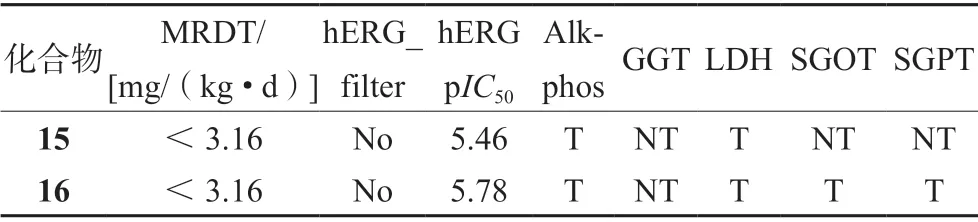
表1 化合物15 和16 在不同模型中的毒性参数预测Tab 1 Predicted toxicity parameters for compounds 15 and 16 in different models

表2 化合物15 和16 对EGFR 受体结合的理论自由焓和HL-60 与DU-145 的IC50 值Tab 2 Theoretical free enthalpies of binding to EGFR receptor and predicted values of inhibition (IC50) for compound 15 and 16
1.2 吲哚类-HDACs 抑制剂
组蛋白的乙酰化状态主要由组蛋白乙酰转移酶和HDACs 共同决定,它们相互拮抗,共同调节基因的表达和细胞周期,对基因的转录具有重要的作用[25]。研究发现,人体内有4 类HDAC 酶,其中Ⅰ、Ⅱ和Ⅳ类HDAC 是金属酶(锌依赖性),需要锌金属离子来触发生物活性[26]。临床研究发现,患有髓样肿瘤和实体瘤的患者体内HDAC 异常增多,因此,HDAC 可能是治疗癌症的靶点[27]。
多个位于表面识别区吲哚核的HDAC 抑制剂已被报道,以HDAC 抑制剂Dacinostat(17,LAQ824)为例,这类抑制剂通常由表面识别区(CAP)、连接区(LINKER)和锌离子结合基团(ZBG)三部分组成(见图7)。体外实验表明,Dacinostat 毒性低并以剂量依赖的方式在结肠癌和肺癌细胞模型中显示出良好的抗癌活性,对细胞总HDAC 的IC50值为32 nmol·L-1,对HDAC1 的IC50值为9 nmol·L-1。目前已经上市的药物有Panobinostat(18,LBH-589,HDACs:IC50=5 nmol·L-1),其能显著抑制多发性骨髓瘤细胞系和多发性骨髓瘤患者新鲜细胞的生长(IC50<40 nmol·L-1)[28]。
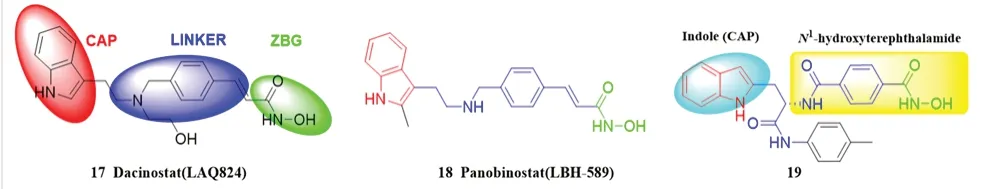
图7 HDAC 抑制剂及其药效团构成Fig 7 HDAC inhibitors and their pharmacophore composition
Wang 等[29]合成了一系列含吲哚帽基的N1-羟基对苯二甲酸类HDACs 抑制剂,其中化合物19对HDAC1(IC50=0.219 μmol·L-1) 和HDAC6(IC50=0.168 μmol·L-1)表现出较好的抑制活性。在体外细胞毒性实验中,化合物19 对宫颈癌细胞Hela(IC50=0.074 μmol·L-1)的抑制效果优于阳性对照Vorinostat(SAHA,IC50=0.131 μmol·L-1),对人白血病HL60 细胞的抑制效果 与SAHA 相 当[(10.80±0.33)μmol·L-1vs(9.37±0.39)μmol·L-1],对人慢性髓原白血病细胞K562、人多发性骨髓瘤细胞U266、人急性骨髓性白血病细胞KG-1 和人乳腺癌细胞MCF-7 细胞株的抑制效果均差于SAHA,这可能是由于化合物19 的跨细胞渗透性差造成的。如图8 所示,化合物的N1-羟基甲酰胺片段的羟基、氮原子、羰基氧原子和吲哚环的氮原子与氨基酸His145、His146、Try308 和Asp104 有6 个氢键相互作用。从立体构型来看,化合物19 的苯环与Phe155 的苯环有π-π堆积作用,这也提高了与HDAC2 结合的活性。

图8 化合物19 与HDAC2 的共晶结构模拟(PDB:4LXZ;锌离子为图中灰色的球体)Fig 8 Co-crystal structure simulation and surface view of compound 19 with HDAC2(PDB:4LXZ,zinc ion is indicated gray at the bottom of the tunnel)
Hati 等[30]发现夹竹桃科和茜草科等天然植物中的抗癌活性成分均含有螺[二氢吲哚-3,3'-吡咯烷]结构,因而对该结构进行修饰改造,发现其具有HDAC2 抑制活性。Hati 等[30]采用一锅法将N-溴代琥珀酰胺介导色胺的Pictet Spengler 氧化环收缩,取代传统的形成四氢-β-咔啉中间体的两步法,大大提高了合成产率(见图9)。在合成的一系列螺[吡咯烷-3,3'-羟吲哚]衍生物中,化合物20 对MCF-7的IC50=(60.96±0.09)μmol·L-1、对非洲青猴肾细胞COS-7 的IC50=(15.42±0.18)μmol·L-1、对人正常乳腺癌上皮细胞MCF10A 的IC50=0.08 μmol·L-1,都有显著的抑制作用。共晶结构模拟图显示,化合物20 与HDAC2 蛋白(4LY1)的His183残基有一个氢键作用和氨基酸Phe155、Phe210 和Leu276 之间存在疏水相互作用(见图9)。
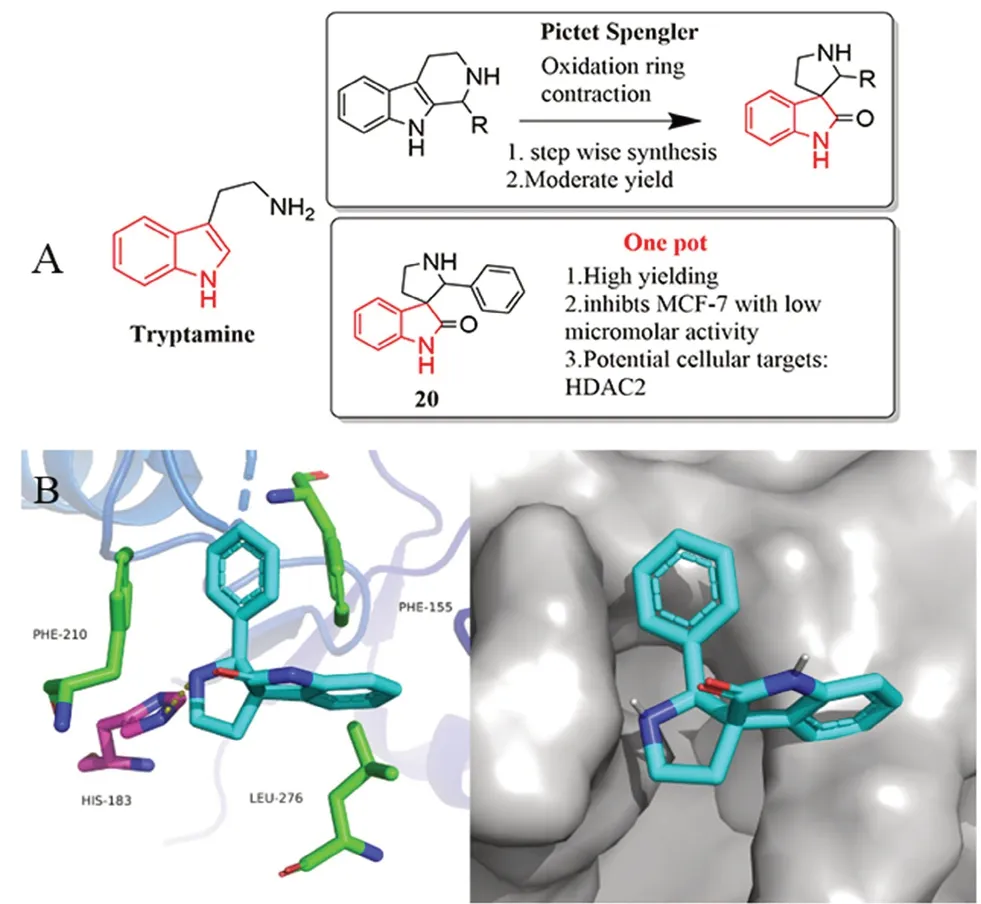
图9 螺[吡咯烷-3,3'-羟吲哚]母核的合成策略及化合物20 与HDAC2(PDB:4LY1)的共晶结构模拟Fig 9 Synthesis strategy of spiro[pyrrolidine-3,3'-oxindole] core and co-crystal structure simulation and surface view of compound 20 with HDAC2(PDB:4LY1)
1.3 吲哚类雌激素受体调节剂
在女性群体中,乳腺癌(breast cancer)是仅次于肺癌的致死性癌症[31],至少70%的乳腺癌患者被归类为雌激素受体(ER)阳性癌症。由于大多数乳腺癌是雌激素依赖性的,因而通常使用雌激素疗法来治疗乳腺癌,这种疗法能有效阻断雌激素与ER 的结合从而减少对乳腺癌细胞的刺激作用[32-33]。选择性雌激素受体调节剂(SERM)是通过与细胞内ER(ER-α或ER-β)结合而降低雌激素作用的化合物。Bazedoxifen(21)和ERA923(22)是两种抗雌激素活性的吲哚衍生物,化合物23 是第三代SERM,对ER-α的亲和力较强[34],化合物22 可以克服Tamoxifen(治疗阳性乳腺癌的代表性SERM)的耐药性[35]。有报道称,2-芳基吲哚衍生物(23)通过靶向ER-α发挥其抗雌激素作用[4]。另有研究表明,1-苄基-3-甲醇(24)吲哚类似物是I3C 活性的1000 倍[36],因此可以证明吲哚N 位的苄基化显著提高了其抗癌活性(见图10)。

图10 有抗雌激素活性的吲哚衍生物的构效关系图Fig 10 Structure activity relationship of indole derivatives with antiestrogenic activity
2 位取代的苯并咪唑结构被证实具有广谱抗癌效果[37],研究发现,苯并咪唑部分中游离的氨基可作为ER-α受体空腔中的氢键供体[38]。Singla 等[39]基于吲哚和苯并咪唑衍生物的结构特征,设计出了一系列吲哚-苯并咪唑类衍生物(见图11)。其中化合物25和26活性最高,化合物25对人乳腺管癌T-47D细胞的IC50为(15.48±0.10)μmol·L-1,对ER 激酶的IC50为(73.61±3.25)nmol·L-1,化合物26 对T-47D 细胞的IC50为(4.99±0.60)μmol·L-1,对ER 激酶的IC50为(80.36±7.02)nmol·L-1;且这两个化合物都是通过改变ER-α的mRNA 和受体蛋白的表达从而阻止人乳腺癌细胞T-47D 的信号传导。
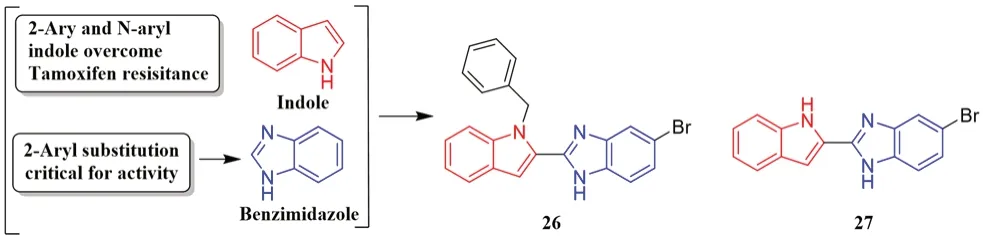
图11 化合物25、26 的设计思路Fig 11 Synthetic strategy for compound 25 and 26
有报道称,苯基取代的二氧八氢氧杂蒽化合物对MCF-7 细胞的IC50值达到了0.02 μmol·L-1[40]。此外,2-芳基吲哚衍生物能靶向抑制ER-α受体,Singla 等[41]将1-苄基吲哚的2 位引入二氧八氢氧杂蒽,合成了一系列吲哚-二氧八氢氧杂蒽类衍生物,并进行了抗增殖活性、细胞毒性和ER-α结合力的筛选,从中挑选出活性较优的化合物27 和28(见图12),两者均表现出对ER-α激酶的抑制和抗增殖作用。化合物27 对T-47D 细胞的IC50为(16.51±0.75)μmol·L-1,对ER-α激酶的IC50为(55±1.97)nmol·L-1;化合物28 对T-47D 细胞的IC50为(17.94±1.0)μmol·L-1,对ER-α激酶的IC50为(16.55±1.95)nmol·L-1。
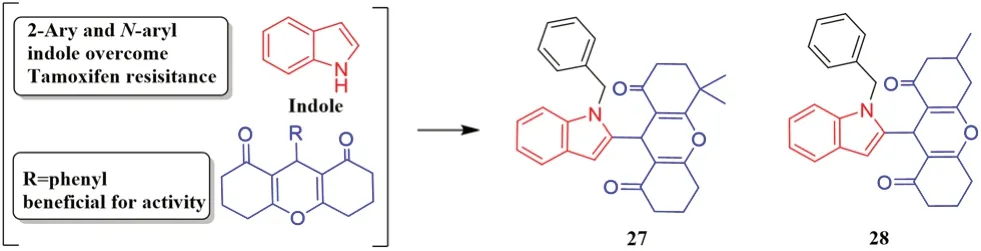
图12 吲哚类ER 受体化合物Fig 12 ER receptor compounds with indole structures
Hendy 等[42]利用生物启发性有机合成(BIOS)和B/C 处位于甾体骨架的开环策略,克服了甾体支架存在的问题,合成了一系列靶向ER-α的仿生雌二醇构型的新型吲哚类支架。在体外细胞毒性实验中,化合物29 和30 表现最佳,可作为针对ER-α药物的新型吲哚类骨架(见图13)。化合物29 对MCF-7 细胞的IC50为(30.63±0.72)μmol·L-1,对T-47D 细胞的IC50为(28.23±0.95)μmol·L-1,对ER-α激酶的IC50为1.76 nmol·L-1;化合物30对MCF-7 细胞的IC50为(30.89±0.96)μmol·L-1,对T-47D 细胞的IC50为(32.96±0.69)μmol·L-1,对ER-α激酶的IC50为3.31 nmol·L-1。

图13 化合物29 和30 的设计策略Fig 13 Synthetic strategy for compound 29 and 30
1.4 吲哚类VEGFR 抑制剂
VEGFR 是胚胎形成、骨骼生长和血管生成最重要的受体之一[43],其与肿瘤、眼内新生血管疾病和其他疾病息息相关。当VEGF 与异常的VEGFR结合后,会产生一系列级联反应,导致肿瘤细胞的无限增殖。目前,临床应用的吲哚类VEGFR激酶抑制剂有西地尼布(31,VEGFR2:IC50<5 nmol·L-1)和莫特沙尼(32,VEGFR1:IC50=2 nmol·L-1;VEGFR2:IC50=3 nmol·L-1;VEGFR3:IC50=6 nmol·L-1,见图14)。
Kim 等[44-45]在白桑树果实中提取分离到了吲哚乙酸衍生物,研究发现,该吲哚乙酸衍生物能抑制VEGFR 并显示出较强的抗癌活性。在无细胞毒性的化合物33 的羧基上引入丁基侧链后,得到化合物34(见图14),在100 μmol·L-1的水平下可有效抑制(52.90±1.88)%的宫颈癌Hela 细胞。有文献报道,增加亲脂性可以提高小分子抑制剂杀死癌细胞的效率,这可能是丁基与VEGFR 结合位点的疏水作用在抗肿瘤活性中起着重要的作用[46]。
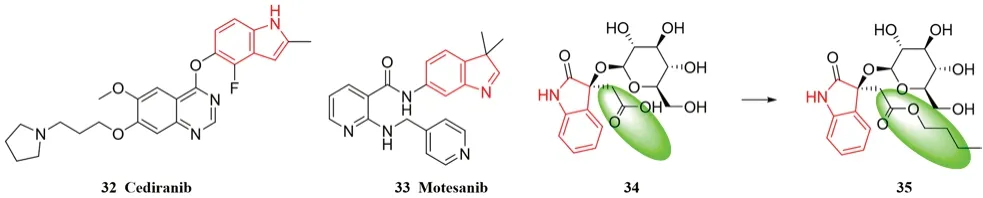
图14 吲哚类VEGFR 抑制剂Fig 14 VEGFR inhibitors with indole structures
1.5 吲哚类SIRT 受体抑制剂
Ⅲ蛋白型赖氨酸去乙酰化酶(SIRT),是一组烟酰胺腺嘌呤二核HDAC 组蛋白去乙酰化酶。SIRT家族包含7 个成员,SIRT1 ~SIRT7[47-48],当SIRT1被激活后可影响下游相关转录因子(如NDRG1)、促凋亡因子(如p53、NF-κb)、修复因子(如Ku70)和叉头转录因子(如FOXOs)的调节,使抑癌基因沉默、增强细胞的修复作用、抑制细胞凋亡、延长细胞寿命[49](见图15)。当SIRT1 过表达时,则会持续触发上述机制,导致恶性肿瘤的产生。
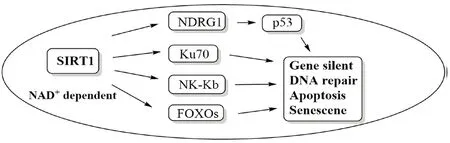
图15 SIRT1 促癌效应机制Fig 15 Mechanism of cancer promoting effect of SIRT1
Panathur 等[50]在吲哚核的2 位引入苄基醚、具有抗癌活性的异恶唑酮衍生物、增加小分子脂溶性的氟原子和三氟甲基,合成了一系列含取代酰胺的吲哚-异噁唑酮杂化物。在体外抗增殖活性实验中,以Gemcitabine 和Suramine 为参照,大多数化合物对人乳腺癌细胞展现出了抑制活性,其中化合物35 和化合物36(见图16B)的抑制效果最佳(见表3),对人乳腺癌细胞的IC50均低于10 μmol·L-1,对SIRT1 激酶的IC50在40 μmol·L-1左右。如图16A 所示,化合物36 的疏水位点朝向Phe297 和Phe273,亲水位点朝向Asp348 和Ile347。
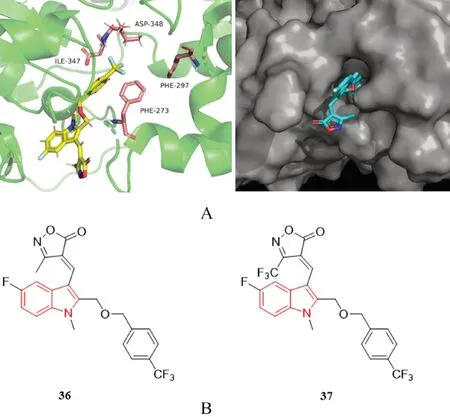
图16 化合物35 与SIRT1 的共晶结构模拟图(PDB:4I5I,A)及吲哚类SIRT 抑制剂(B)Fig 16 Co-crystal structure simulation and surface view of compound 35 with SIRT1(PDB:4I5I,A)and SIRT inhibitors with indole structures(B)
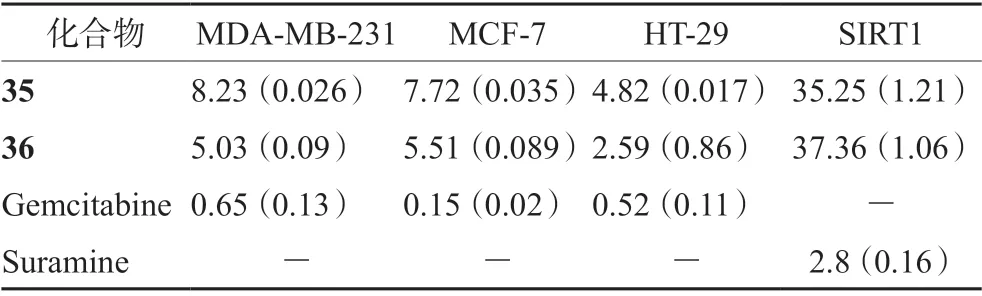
表3 化合物35 和36 对各类癌细胞的IC50 值(μmol·L-1)Tab 3 IC50 value of compound 35 and 36 on various cancer cells(μmol·L-1)
1.6 吲哚类拓扑酶受体药物
拓扑酶是细胞核中的一种重要酶类,分为拓扑酶Ⅰ(TopoⅠ)和拓扑酶Ⅱ(TopoⅡ)两种类型。这两类酶对DNA 的转录、复制以及基因表达起着重要作用。拓扑酶抑制剂是一种靶向抗癌药物,其通过抑制拓扑酶的活性从而阻止肿瘤细胞的快速增殖,达到抗癌的效果。有文献报道,β-咔啉9 号位N 原子上的取代基被证明是拓扑酶抑制剂产生细胞毒性和与DNA 结合活性的关键,N 原子上的取代基与DNA 结合能力越强,细胞毒性也就越强[51];双羟基吡咯环是诱导DNA 交联的抗肿瘤活性药效团,吡咯环上的羟甲基可以形成两个亲电中心,通过SN1 亲电反应与DNA 双链交联[52],阻断肿瘤细胞中DNA 的复制,杀死肿瘤细胞。
因此,Eswar 等[53]将β-咔啉基团和双(羟甲基)吡咯两个药效基团整合,合成了一系列双(羟甲基)吲哚并[6,7-b]吲哚杂化物(见图17)。用TopoⅠ抑制剂Irinotecan 和TopoⅡ抑制剂Etoposide作为阳性对照,72 h 后化合物37 在0.5 μmol·L-1对TopoⅠ和TopoⅡ有显著的双重抑制作用。此外,化合物37 在异种移植肿瘤模型中具有广谱的抗肿瘤活性,能够有效抑制多种NSCLC 系列细胞且能有效杀死对吉非替尼产生耐药性的PC-9/gef B4 细胞[IC50=(1.58±0.33)μmol·L-1]。
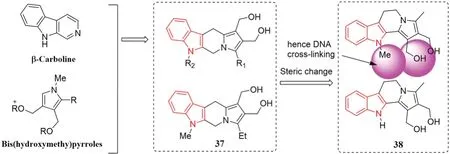
图17 吲哚类拓扑酶受体抑制剂的设计思路Fig 17 Synthesis strategy of indole topo enzyme receptor inhibitors
Chen 等[54]对双(羟甲基)吡咯并[6,7-b]吲哚杂化物进行了空间改造,发现化合物38(见图17)对TopoⅡ的抑制效果最佳。在体外细胞毒性实验中,化合物38 表现出较为广泛的抗肿瘤活性,对SCLC 细胞的作用尤为显著[人急性淋巴白血病细胞CCRF-CEM:IC50=(1.02±0.32 μmol·L-1,人结肠癌细胞HTC-116:IC50=(2.56±0.27)μmol·L-1,人前列腺癌细胞PC-3:IC50=(3.53±0.31)μmol·L-1,人大细胞肺癌H460:IC50=(3.46±0.34)μmol·L-1,人肺癌细胞H211:IC50=(0.22±0.03)μmol·L-1]。在体内抗肿瘤活性实验中,化合物38对SCLC H526 裸鼠移植瘤的生长速度的抑制优于顺铂和依托泊苷,与伊立替康相当。与化合物37 不同,化合物38 在吲哚的N 原子处引入甲基后,虽增加了与DNA 的交联,但减小了Topo Ⅱ的抑制作用。计算机分析表明,吲哚N 位的甲基影响了双羟基的扭转角,因而更有利于DNA 链间的交联。
2 总结和展望
近年,吲哚骨架被广泛用于抗肿瘤活性研究,研究者们针对不同靶点设计了许多吲哚类抗癌药物。这些药物大多展现出良好的抗肿瘤活性,但在临床上也表现出不同程度的不良反应和药物耐受性等问题。因此,合成选择性高、毒副作用低和克服耐药问题的新型吲哚类先导化合物具有十分广阔的前景。
猜你喜欢
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:53:38
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
抗癌之窗(2020年1期)2020-05-21 10:18:10
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
特别健康(2018年9期)2018-09-26 05:45:26
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:08
合成化学(2015年10期)2016-01-17 08:56:26
