
赖氨酸磷酸氢钙颗粒现行质量标准存在的问题及改进建议
2022-08-08 03:21李春焕巩丽萍薛维丽张乃斌杭宝建咸瑞卿
食品与药品 2022年4期
李春焕,巩丽萍,石 峰,薛维丽,张乃斌,杭宝建,咸瑞卿
(山东省食品药品检验研究院 国家药品监督管理局仿制药研究与评价重点实验室 山东省仿制药一致性评价工程技术研究中心,山东 济南 250101)
赖氨酸磷酸氢钙颗粒是由盐酸赖氨酸、磷酸氢钙与辅料制成的复方制剂,临床主要用于促进幼儿生长发育及儿童和孕妇钙质的补充[1-4]。在国家评价性抽验工作中,我们通过对该品种的质量标准进行研究,发现现行中国药典、美国药典、英国药典、欧洲药典、日本药局方中均收载有盐酸赖氨酸和磷酸氢钙原料药,但均未收载该制剂。目前,我国有16家具备批准文号的生产企业,其中15家执行国家标准WS-10001-(HD-0389)-2002,1家执行注册标准YBH00102018。在本次国家评价性抽验工作中,我们对全国范围内抽取的167批赖氨酸磷酸氢钙颗粒进行了法定标准检验和探索性研究工作,发现现行质量标准中赖氨酸鉴别专属性差、磷酸盐与钙盐的鉴别反应灵敏度低、干燥失重检查和溶出度检查缺失、赖氨酸含量测定方法专属性差及限度设置不合理等问题。为改进和完善赖氨酸磷酸氢钙颗粒的质量标准,我们通过实验分析和比较,对“赖氨酸鉴别”、“磷酸盐与钙盐的鉴别反应”及“赖氨酸含量测定方法”进行了修改,并增订了“干燥失重检查”和“溶出度检查”。
1 仪器与试药
1.1 仪器
Agilent 1260 Infinity II高效液相色谱仪(美国安捷伦科技有限公司);Mettler Toledo XSE205DU电子天平(瑞士梅特勒-托利多公司)
1.2 试药
盐酸赖氨酸对照品(中国食品药品检定研究院,批号:140673-202010,含量:100.0 %);甲醇、乙腈为色谱纯,水为Millipore Q-POD®制备的纯化水,其他试剂均为分析纯。
2 赖氨酸鉴别
2.1 存在的问题
现行质量标准均采用茚三酮与α-氨基酸加热反应产生蓝紫色物质的原理来鉴别赖氨酸,该方法的专属性不强,无法区别赖氨酸与其他氨基酸,需进一步建立专属性强、灵敏性高的赖氨酸鉴别方法。
2.2 方法改进
采用柱前衍生-高效液相色谱法(HPLC)对赖氨酸进行鉴别。
2.2.1 溶液制备
2.2.1.1 对照品溶液制备 精密称取盐酸赖氨酸对照品12 mg,置100 ml量瓶中,加水约30 ml,超声5 min,用水稀释至刻度,摇匀,即得。
2.2.1.2 常见氨基酸混合对照品溶液制备 精密称取盐酸半胱氨酸14.40 mg,酪氨酸21.15 mg,异亮氨酸15.20 mg,盐酸组氨酸24.55 mg,甲硫氨酸18.00 mg,苯丙氨酸22.30 mg,亮氨酸16.50 mg,苏氨酸21.85 mg,缬氨酸15.00 mg,丝氨酸19.95 mg,天冬氨酸16.85 mg,盐酸赖氨酸22.95 mg,谷氨酸18.45 mg,精氨酸25.45 mg,丙氨酸10.11 mg,脯氨酸16.93 mg,盐酸羟赖氨酸14.31 mg,甘氨酸9.59 mg,羟脯氨酸19.38 mg,盐酸鸟氨酸22.85 mg,分别置50 ml量瓶中,加水溶解并稀释至刻度,摇匀,分别取2.5 ml,置同一25 ml量瓶中,制成20种氨基酸的混合对照品溶液。
2.2.1.3 供试品溶液制备 精密称取样品120 mg(相当于盐酸赖氨酸12 mg),置100 ml量瓶中,加水约30 ml,超声5 min,用水稀释至刻度,摇匀,滤过,即得。
2.2.1.4 衍生试剂制备 取2, 4-二硝基氟苯(凝点为25~28 ℃)2 ml,加乙腈至100 ml,摇匀,即得(需避光保存)。
2.2.2 衍生方法 取对照品溶液和供试品溶液各1 ml,分别置10 ml量瓶中,加入0.5 mol/L碳酸氢钠溶液1 ml及衍生试剂0.2 ml,混匀,置暗处,60 ℃水浴放置60 min,取出放冷至室温,加pH 7.0磷酸盐缓冲液(取磷酸二氢钾0.68 g,加0.1 mol/L氢氧化钠溶液29.1 ml,加水稀释至100 ml,即得)稀释至刻度,摇匀,取10 μl注入高效液相色谱仪。
2.2.3 色谱条件 流动相:甲醇-0.05 mol/L醋酸钠缓冲液(用稀醋酸调节pH至6.8±0.05)(65:35);流速:1 ml/min;色谱柱:Thermo BDS HYPERSIL C18(250 mm×4.6 mm,5 μm),Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm;150 mm×4.6 mm,5 μm);柱温30 ℃;检测器:紫外检测器,检测波长:360 nm。
2.3 结果
2.3.1 拟定标准 供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
基于卸点填埋工艺与暴露面积的异味控制,提出导入“面量比”的绩效考核方式,即每天作业面积与垃圾量有合理比例关系,具体评价指标如表4所示。
2.3.2 赖氨酸鉴别结果 167批供试品溶液主峰的保留时间均与对照品溶液主峰的保留时间一致,合格率100 %。
3 磷酸盐与钙盐的鉴别
3.1 存在的问题
WS-10001-(HD-0389)-2002标准中磷酸盐与钙盐的鉴别反应遵循中国药典2020年版四部通则0301,其对磷酸盐鉴别反应(1)描述如下:取供试品的中性溶液,加硝酸银试液,即生成浅黄色沉淀;分离,沉淀在氨试液或稀硝酸中均易溶解[5]。
首先,调节溶液为中性后,因磷酸氢钙在中性水溶液中不溶,因此滤液中钙离子和磷酸氢根离子极少,导致鉴别反应不灵敏;其次盐酸赖氨酸中的氯离子加硝酸银试液后会产生大量白色氯化银沉淀,生成浅黄色磷酸银沉淀的现象不明显;最后取沉淀加氨试液全溶,但加稀硝酸溶液后不能完全溶解,鉴别反应不灵敏,以上反应现象见图1。因此现行赖氨酸磷酸氢钙颗粒质量标准中的磷酸盐和钙盐的鉴别方法灵敏度低,可行性差,鉴别现象不易观察,需对其进行修订。

图1 磷酸盐鉴别反应(1)现象
3.2 改进的方法
取本品的细粉适量(约相当于磷酸氢钙50 mg),加水20 ml,充分振摇,离心过滤,取沉淀,加稀硝酸5 ml和水20 ml溶解,溶液显钙盐和磷酸盐的鉴别反应。
3.3 结果与分析
采用修订后的方法对国内10家生产企业的167批样品进行磷酸盐和钙盐的鉴别,结果均呈正反应,反应现象明显,易于判断。
本方法取样品加适量水溶解,离心过滤,可除去盐酸赖氨酸,避免了氯离子对磷酸盐鉴别的干扰。沉淀主要为磷酸氢钙,分离后用适量稀硝酸溶解,进行磷酸盐和钙盐的鉴别,方法简单,现象明显,易于观察,灵敏度高。
4 盐酸赖氨酸含量测定
4.1 存在的问题
现行质量标准采用紫外-可见分光光度法(茚三酮比色法)测定赖氨酸含量,企业调研及实际检验工作中均发现,该方法受试剂、温度、反应时间等因素影响较大,专属性不强,且限度范围设置过宽(85.0 %~115.0 %),测定结果不能客观准确地反映产品中的真实含量。
4.2 改进的方法
通过参考文献资料[6-10],将赖氨酸含量测定方法修订为柱前衍生-HPLC法,按2.2项下步骤进行衍生化并进样测定。该方法专属性强,准确度高。
4.2.1 耐用性考察 选择不同型号色谱柱进行耐用性考察,结果见表1。使用不同厂家的不同规格色谱柱,盐酸赖氨酸色谱峰保留时间、理论板数符合要求,表明色谱条件耐用性良好。

表1 盐酸赖氨酸含量测定耐用性考察结果
4.2.2 专属性 空白辅料在对照品主峰保留时间处未有色谱峰出现;相同条件下处理亮氨酸等20种氨基酸,仅亮氨酸保留时间与赖氨酸较为接近,两峰分离度为2.0;因此本方法中空白辅料、衍生化试剂和实验过程中用到的其他试剂及其他氨基酸对赖氨酸的鉴别均无干扰,该方法对鉴别盐酸赖氨酸具有较好的专属性。色谱图见图2~4。
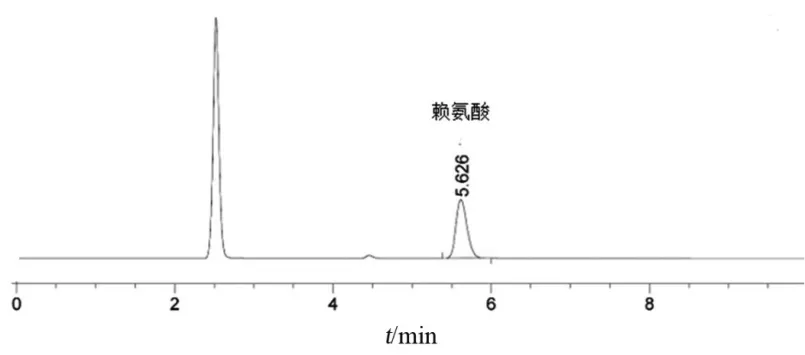
图2 典型样品色谱图
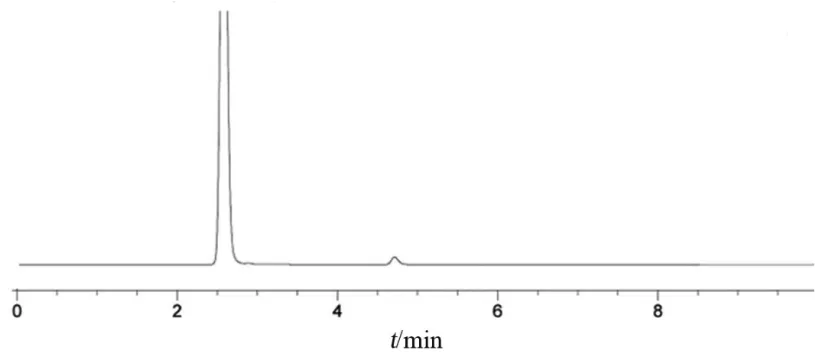
图3 空白辅料色谱图

图4 20种氨基酸混合对照品色谱图
4.2.3 线性及范围 精密称取对照品60.02 mg,置5 ml量瓶中,加水溶解并稀释至刻度,摇匀,制成每1 ml约含12 mg的盐酸赖氨酸对照品储备液。分别精密量取上述对照品储备液10,20,50,100,150,200,300,400 μl,置10 ml量瓶中,加水稀释至刻度,摇匀,作为对照品系列溶液。按2.2项下方法,分别取对照品系列溶液注入液相色谱仪,测定,以浓度为横坐标,峰面积为纵坐标,进行线性回归。回归方程:Y=10.238X-18.317,r=1.0000。结果表明,在12~480 μg/ml范围内盐酸赖氨酸浓度与峰面积线性关系良好。
4.2.4 精密度 精密吸取盐酸赖氨酸对照品溶液(0.1200 mg/ml)10 μl,注入液相色谱仪,连续进样6次,测其峰面积,计算得RSD=0.19 %,表明仪器精密度良好。
不同分析人员,在不同日期,取同厂家同批样品(批号:20200103),按2.2.1.3项下方法制备供试品溶液,按2.2.2项下方法进行样品衍生,按2.2.3项下色谱条件,用不同仪器进样测定,平均含量为97.62 %,RSD=1.22 %(n=12),表明方法中间精密度良好。
4.2.5 稳定性 精密吸取供试品溶液10 μl,注入液相色谱仪,分别于4 h、20 h及24 h进样一次,测定盐酸赖氨酸色谱峰的峰面积,计算峰面积的RSD,观察检测过程中待测成分的稳定性。结果,峰面积RSD为1.04 %,表明24 h内供试品稳定性良好,能满足测定需要。
4.2.6 重复性 取样品约120 mg,精密称定,按2.2.1.3项下方法,平行制备6份供试品溶液。精密吸取上述供试品溶液10 μl,注入液相色谱仪,以外标法计算盐酸赖氨酸。结果,峰面积RSD为1.06 %,表明方法的重复性良好。
4.2.7 回收率 按颗粒剂处方制备不含盐酸赖氨酸的空白颗粒(含磷酸氢钙10 g,蔗糖80 g)。分别称取9份混合均匀的空白颗粒适量(约110 mg),分为3组,置100 ml量瓶中,每组分别加入对照品储备液(精密称取对照品60.04 mg,置5 ml量瓶中,加水溶解并稀释至刻度,摇匀,作为对照品储备液)0.5,1.0,1.5 ml,加水溶解并稀释至刻度,摇匀,制得3个浓度水平的9份供试品溶液,按2.2.1.3项下方法处理,按2.2.2项下方法进行样品衍生,按2.2.3项下色谱条件进样测定,结果见表2。结果表明,测定方法的回收率良好。
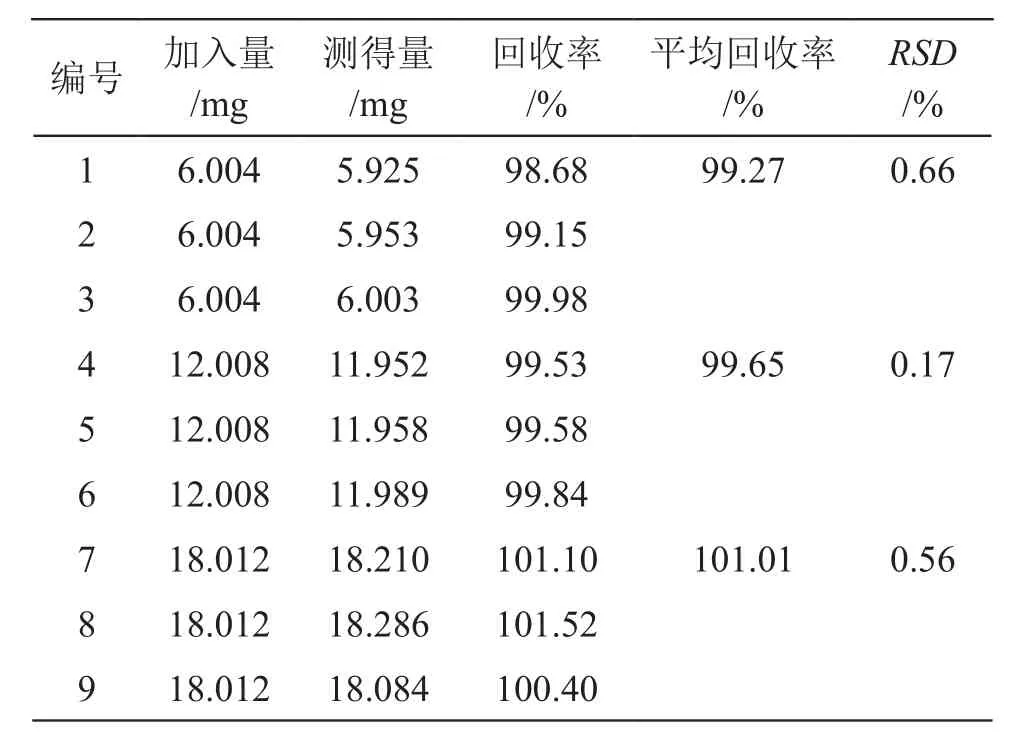
表2 加标回收率考察结果
4.2.8 检出限与定量限 精密量取对照品溶液(0.1201 mg/ml)1 ml,置100 ml量瓶中,加水稀释至刻度,混匀,精密量取3 ml和10 ml,分别置100 ml量瓶中,加水稀释至刻度,混匀,分别得浓度为0.036,0.12 μg/ml的对照品溶液。衍生化后取10 μl进样测定,信噪比分别为3.1和10.4,得盐酸赖氨酸的检出限和定量限分别为0.036,0.12 μg/ml(以衍生前盐酸赖氨酸计)。
4.3 结果与分析
分别根据现行质量标准W S-1 0 0 0 1-(HD-0389)-2002(UV法)及拟定标准(柱前衍生-HPLC)测定10家生产企业的167批样品,结果显示,拟定方法专属性强、重现性好,能有效反映产品质量,各批次样品检测结果均在90.0 %~110.0 %范围,因此限度修订为“90.0 %~110.0 %”较为合理。法定标准的检测结果显示其含量分布范围较改进后方法宽,且现行标准方法采用紫外-可见分光光度法,专属性不强,因此建议采用灵敏度高、选择性好的柱前衍生HPLC法测定样品中赖氨酸的含量。
5 增订干燥失重检查
赖氨酸磷酸氢钙颗粒为含糖颗粒,根据《中国药典》2020年版制剂通则颗粒剂项下要求需检查干燥失重,现行国家标准WS-10001-(HD-0389)-2002无干燥失重检查项,注册标准YBH00102018收载了干燥失重检查。因此我们对收集的所有颗粒剂样品,按《中国药典》2020年版四部“干燥失重”检查法测定颗粒剂干燥失重,考察各企业工艺稳定性,发现1批不合格的样品。因此增订赖氨酸磷酸氢钙颗粒“干燥失重”的检查是有必要的。
根据探索性研究,我们拟增订赖氨酸磷酸氢钙颗粒干燥失重测定方法:取本品,在80 ℃减压干燥至恒重,减失重量不得过2.0 %[5]。
6 增订溶出度检查
6.1 存在的问题
溶出度是口服固体制剂的重点关注项目,是体现药品有效性的重要指标,溶出曲线反映了药品的溶出速率[5,11]。赖氨酸磷酸氢钙颗粒为混悬颗粒,需进行溶出度检查,但现行国家质量标准WS-10001-(HD-0389)-2002中无溶出度检查项,注册标准YBH00102018虽收载了溶出度检查方法,但存在溶出条件不合理、测定方法专属性不强等缺点。此次我们探索赖氨酸磷酸氢钙颗粒的溶出介质、溶出方法及溶出限度,建立合适的溶出条件及检测方法,增订溶出度检查项。
6.2 改进的方法
拟增订赖氨酸磷酸氢钙颗粒溶出度测定方法:取本品,照溶出度与释放度测定法[5]以盐酸溶液(9→1000)900 ml为溶出介质,转速为每分钟50转,依法操作,经45 min时,取溶液适量,滤过,取续滤液适量,备用。
6.2.1 盐酸赖氨酸溶出量测定及限度要求 精密量取上述滤液2 ml,加水定容至10 ml,摇匀,作为供试品溶液。精密量取上述供试品溶液1 ml,照改进后的含量测定方法,依法测定,计算每袋溶出量,限度为标示量的80 %。
6.2.2 磷酸氢钙溶出量测定及限度要求 精密量取上述滤液5 ml,置50 ml量瓶中,加5 %镧溶液(取氧化镧6.6 g,加盐酸10 ml使溶解,加水稀释至100 ml,摇匀)1 ml,加0.1 mol/L盐酸溶液稀释至刻度,摇匀,作为供试品溶液。取磷酸氢钙对照品约25 mg,精密称定,置50 ml量瓶中,加0.1 mol/L盐酸溶液使溶解并稀释至刻度,摇匀,精密量取4.5 ml、5.0 ml、5.5 ml,分别置50 ml量瓶中,各加5 %镧溶液1 ml,分别用0.1 mol/L盐酸溶液稀释至刻度,摇匀,作为对照品溶液。取供试品溶液与对照品溶液,照原子吸收分光光度法(中国药典2020年版四部通则0406第一法)[5],在422.7 nm的波长处测定,计算每袋的溶出量,限度为标示量的80 %。
6.3 方法学研究
6.3.1 方法的选择 注册标准YBH00102018采用篮法,但未收集到该企业的样品,未对该方法的适用性进行研究。部分其他企业由于粒度较小,采用篮法时,颗粒或粉末易从篮网漏出,不便于操作,因此采用桨法。
6.3.2 溶出介质的选择 采用桨法,转速为50转/min,分别选择水、0.1 mol/L盐酸溶液、醋酸盐缓冲液(pH 4.5)、磷酸盐缓冲液(pH 6.8)为溶出介质,体积均为900 ml,考察赖氨酸磷酸氢钙颗粒中盐酸赖氨酸和磷酸氢钙的溶出行为。
由于盐酸赖氨酸易溶于水,磷酸氢钙在水中不溶、在稀盐酸或稀硝酸中易溶。因此,盐酸赖氨酸在4种溶出介质中均能较快溶出(见图5)。磷酸氢钙在水、磷酸盐缓冲液(pH 6.8)中,60 min时溶出度均小于10 %,在醋酸盐缓冲液(pH 4.5)中60 min时溶出度小于45 %,在0.1 mol/L盐酸溶液中30 min时溶出度超过80 %(见图6)。以上结果显示赖氨酸和磷酸氢钙在0.1 mol/L盐酸溶液中溶出良好,故选择900 ml的0.1 mol/L盐酸溶液作为溶出介质。

图5 不同溶出介质中盐酸赖氨酸溶出曲线
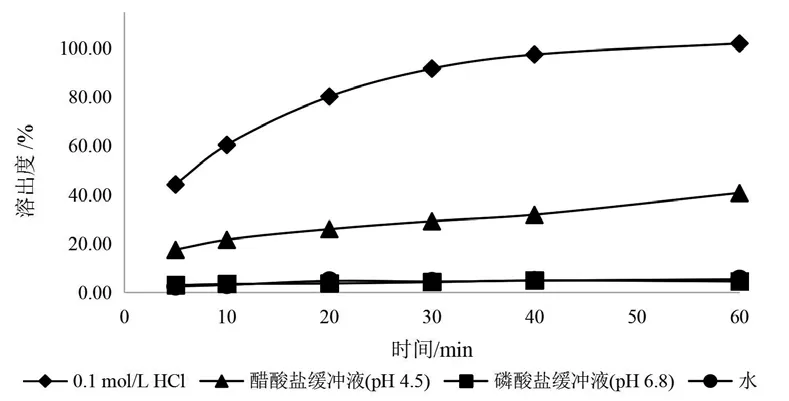
图6 不同溶出介质中磷酸氢钙溶出曲线
6.3.3 转速的选择 采用桨法,以900 ml 0.1mol/L盐酸溶液为溶出介质,比较样品在75转/min和50转/min两种转速下的溶出行为(见图7、图8)。结果显示在75转/min条件下盐酸赖氨酸和磷酸氢钙的溶出速度过快,20 min时溶出量均超过90 %;在50转/min转速下即可较快溶出,所以选择50转/min进行试验。
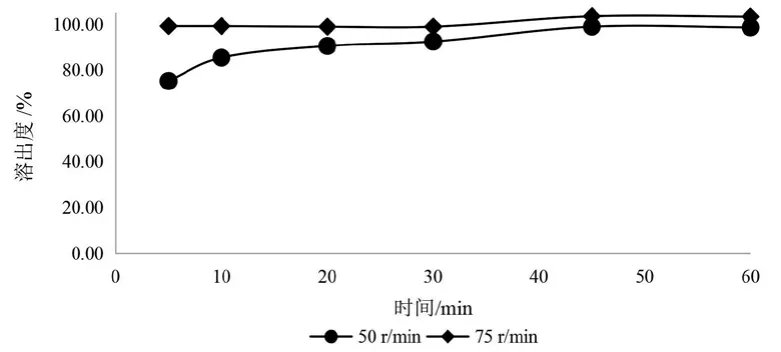
图7 不同转速下盐酸赖氨酸溶出曲线
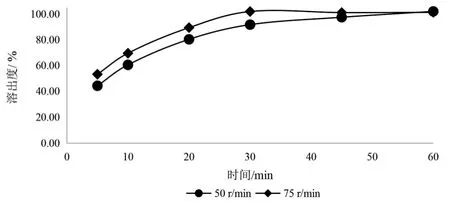
图8 不同转速下磷酸氢钙溶出曲线
6.4 结果与分析
参考167批样品的检测数据,我们将盐酸赖氨酸和磷酸氢钙的溶出度限度均设定为不低于标示量的80 %。用拟定方法测定167批颗粒剂的溶出度,合格率为100 %。对检验结果进行汇总分析后发现,167批颗粒剂样品的盐酸赖氨酸平均溶出度为98.5 %,RSD为2.38 %;磷酸氢钙平均溶出度为92.9 %,RSD为2.82 %。各生产厂家盐酸赖氨酸的溶出度均高于磷酸氢钙,与盐酸赖氨酸极易溶解有关。不同生产厂家之间两种成分的溶出度离散程度均较大,因此有必要在标准中增订溶出度项,确保产品质量的稳定性。
7 小结
质量标准是药品质量控制的依据,检验项目设置的合理性与方法的科学性、先进性体现了药品的内在质量。通过对赖氨酸磷酸氢钙颗粒进行国家评价性抽验工作,提示赖氨酸磷酸氢钙颗粒现行质量标准不能满足质量控制的要求,建议对现行质量标准进行统一和完善,修订鉴别项目、增订缺失的检验项目、采用专属性强、灵敏度高的鉴别和含量测定方法,设置合理的含量限度。
猜你喜欢
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
药品评价(2021年17期)2021-11-06
湖南饲料(2021年4期)2021-10-13
智富时代(2018年6期)2018-08-06
智富时代(2018年6期)2018-08-06
时尚育儿(2018年8期)2018-05-14
中国高新技术企业(2015年24期)2015-06-25
药学研究(2012年2期)2012-10-25
药学研究(2012年3期)2012-08-15
