
页岩油在无机矿物表面赋存运移特征的分子动力学模拟
2022-07-28 09:25:48程林松曹仁义贾志豪
西安石油大学学报(自然科学版) 2022年4期
黄 涛,程林松,曹仁义,王 鹏,贾志豪
(中国石油大学(北京) 石油工程学院,北京 102249)
引 言
页岩油藏矿物组成复杂,常见的无机矿物成分有石英、长石、碳酸盐矿物和黏土矿物等[1-3]。不同矿物的亲油性不同,会对页岩油的赋存和运移产生不同程度的影响,因此大量学者研究了不同矿物对页岩油赋存运移的影响。宁方兴等[4]运用扫描电镜等手段分析了济阳坳陷主力岩相石英、方解石和黏土矿物等矿物中页岩油的赋存状态;王民等[5]通过有机地球化学等方法研究了不同矿物组成下吸附油和游离油的含量;CUI等[6]基于高斯混合理论建立了页岩油赋存状态的模型,研究了有机质和无机质矿物对页岩油吸附状态的影响;ZHANG等[7]通过实验研究了温度和压力对不同矿物表面润湿性的影响。上述学者多基于实验和理论推导矿物组成对页岩油赋存状态的影响,缺少了对微观现象的描述和机理解释。
分子动力学模拟可以从纳米级别上模拟油气的赋存及运移,研究页岩油与储层的微观作用机制,因此也有学者运用此方法研究页岩油与不同矿物表面的相互作用机理,ZHAO等[8-9]运用分子动力学的方法研究了不同矿化度下方解石不同表面对油分子运动的影响;WANG等[10]利用分子动力学模拟研究了石英纳米孔中正辛烷的静态性质和压力驱动下的流动行为;吴春正等[11]运用分子动力学模拟方法研究了混合烃在二氧化硅和石墨狭缝中的密度分布特征及温度对有机质狭缝内烃分子吸附的影响;LUAN等[12]运用分子动力学模拟研究了石英矿物中二氧化碳驱油的机理;TIAN等[13]利用分子动力学模拟研究了多组分油混合物在高岭石孔隙中的吸附特性。但这些学者多研究一到两种矿物中原油的赋存运移行为,不能完整地体现页岩储层无机矿物中的页岩油的赋存运移规律。
本文首先通过构建石英、钠长石、方解石和高岭石4种矿物模型来分别表征页岩储层中的陆源碎屑岩、碳酸盐岩及黏土矿物,然后通过构建甲苯和己烷两种组分代表页岩油,最后运用分子动力学模拟了页岩油在不同矿物表面的吸附扩散,从微观上描述了不同矿物对页岩油赋存运移的规律并解释了机理,对页岩油藏的勘探开发有重要的意义。
1 模型的构建
1.1 岩石和流体模型的构建
根据文献中的数据,高岭石和钠长石属于三斜晶系,高岭石单位晶胞的空间群为P1[14],钠长石单位晶胞的空间群为C1[15];方解石和石英属于三方晶系[16],方解石单位晶胞的空间群为R3C,石英单位晶胞的空间群为P3221。矿物模型的具体晶胞参数见表1。根据表1中的数据,运用Material Studio 8.0软件建立晶胞模型。4种矿物的晶胞如图1所示。

表1 矿物模型的晶胞参数

图1 4种矿物晶胞图
矿物存在不同的晶面,不同晶面的稳定性不同,并对孔隙内流体有着不同的影响。迄今为止对方解石的{104}晶面[17 -18]和其他3种矿物的{001}晶面[19]开展的研究较多,所以本文研究基于方解石的{104}面和其他3种矿物的{001}晶面进行。
页岩油中的饱和烃和芳香烃的占比较高,一般大于80 %,因此建立以甲苯为代表的芳香烃模型和以己烷为代表的饱和烃模型,并且由于甲苯和己烷结构的对称性不同,也可以代表页岩油中的非极性物质与极性物质[20]。甲苯和己烷的模型如图2所示。
图2 流体模型
1.2 计算模型的构建
通过构建平板状的计算模型来模拟石油组分在矿物表面的润湿,计算模型构建如下:(1)首先为了消除模型边界效应,采用周期性边界条件;(2)其次在空间群为P1下建立4a×2b×1c的矿物超晶胞模型;(3)最后将30个甲苯或者己烷分子组成的液滴模型放在不同矿物的超晶胞模型上,构成计算模型。计算模型如图3所示。
图3 计算模型
2 模拟过程
本文中矿物模型和流体模型的力场均采用COMPASS力场,这是一个将从头算法和实验参数相结合的力场,能够准确地预测无机物和有机物的相关性质[21]。其总势能的函数公式为:
(1)
式中:前5项为成键形式的能量,分别表示键伸缩、键角变化、二面角变化、交叉项和离平面的弯曲势变化引起的能量改变的加和,b,θ,φ,χ分别为键长、键角、二面角和离平面振动的角度;最后2项表示非键相互作用,分别为静电相互作用和范德华力相互作用,其中采用L-J9-6势能模型计算范德华力和静电力的相互作用[22],其计算方法为:
(2)
(3)
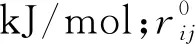
计算模型采用Smart算法计算15 000步找到最优结构。静电作用势能计算选择Ewald求和法,分子之间的范德华力计算采用Atom based算法,其截断半径为1.55 nm。
在动力学计算中,采用NVT系综,控温方法采用Nose-Hoover扩展系统法,总模拟时长500 ps,步长为1 fs。其他参数与前文一致。
3 结果与讨论
利用软件模拟分析在温度为298 K条件下甲苯和己烷在4种矿物表面运移扩散的规律。
3.1 润湿接触角
结构优化后的甲苯和己烷在4种矿物表面的润湿角如图4和5所示,从图中可以发现,甲苯在除高岭石之外的3种矿物表面也会向球形转变,甲苯在高岭石表面会平铺开来。通过拟合甲苯在4种矿物表面的接触角可得石英的接触角约为92.7°,方解石的接触角约为80.7°,钠长石的接触角约为68.5°。己烷在4种矿物的表面都会聚集起来向球形转变。通过拟合己烷在4种矿物表面的接触角可得石英的接触角约为89.7°,方解石的接触角约为78.5°,钠长石的接触角约为63.5°,高岭石的接触角约为61.3°。接触角越小,矿物对该液体的润湿性越强,亲油性越好,因此4种矿物的亲油性排序由高到低为:高岭石>钠长石>方解石>石英。这与张永超[23]和吴春正[24]的结果基本类似,证明了模型的正确性。

图4 甲苯在4种矿物表面的润湿接触角

图5 己烷在4种矿物表面的润湿接触角
3.2 相对密度分布
分子动力学平衡过后,甲苯和己烷在4种矿物表面的分布如图6所示。从图6中可知,甲苯和己烷会在矿物表面重新分布,但不同矿物表面的甲苯和己烷分子的密度不同,其中甲苯和己烷都会在高岭石、钠长石和方解石矿物表面形成一个明显的聚集层,但甲苯则在远离石英矿物表面的真空层形成聚集,己烷会在石英矿物的表面和远离矿物表面的真空层中各形成一个吸附层。由前文的分析可知,这是由于不同矿物表面的亲油性不同造成的,石英的亲油性最差,因此矿物表面的油分子吸附得少,多余的油分子会运动到远离矿物的真空层中;高岭石亲油性最强,因此表面的油分子密度最大。
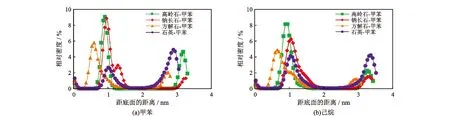
图6 甲苯和己烷在4种矿物表面的相对密度分布
3.3 速度剖面
甲苯和己烷在4种矿物表面的速度剖面图分别如图7和图8所示,其中速度分为沿X轴、Y轴和Z轴3个方向的分速度。从图中可知,甲苯在钠长石和方解石表面的速度峰值出现在远离矿物表面的真空层中,石英和高岭石表面的甲苯运动速度较快,而在真空层中的甲苯运动的速度相对较小,尤其是石英真空层中甲苯运动速度几乎为0;己烷在石英矿物的表面运动速度很快,在远离矿物表面的真空层中运动速度只有矿物表面速度的一半左右,而己烷在另外3种矿物表面的表现正好相反,其速度的峰值出现在真空层中。结合前文分析的相对密度分布可知,这是由于分子动力学平衡后甲苯和己烷在高岭石、钠长石和方解石表面形成聚集层,所以在真空层中的甲苯和己烷会加速运动到矿物表面,因此钠长石和方解石真空层中的甲苯以及高岭石、钠长石和方解石真空层中的己烷的运动速度较大。甲苯在高岭石矿物表面结构优化后就平铺在矿物表面,并且表面的甲苯分子较多,所以会在高岭石表面出现速度剖面的峰值;甲苯和己烷在石英矿物表面密度较小,真空层中密度较高,因此表面的甲苯和己烷加速运动到真空层中,所以石英矿物表面的甲苯和己烷速度较大。

图7 甲苯在4种矿物表面的速度剖面图

图8 己烷在4种矿物表面的速度剖面图
3.4 相互作用机制
模拟计算的4种矿物与甲苯和己烷之间的相互作用能如图9所示,其中静电能是指由于分子中的原子带有部分电荷而引起静电吸引或排斥作用,主要描述粒子之间的库仑力。图中能量正负值代表吸附和排斥作用,正值代表排斥,负值代表吸附。从图中可知,甲苯和己烷与4种矿物之间的相互作用总能量均为负值,说明甲苯和己烷与4种矿物之间为吸附关系。甲苯和己烷与4种矿物的相互作用总能量按从大到小排序为高岭石>钠长石>方解石>石英,这与前文的4种矿物的亲油性的关系一致,表明矿物表面与液体分子之间的作用力的大小决定了矿物的亲油性,矿物表面与油分子的相互作用力越强,矿物越亲油。4种矿物与甲苯之间的静电能大于范德华能, 表明4种矿物与甲苯之间吸附时静电力起主要作用。4种矿物吸附己烷时,范德华能的绝对值均大于静电能的绝对值,说明4种矿物吸附己烷时范德华能起主要作用;除了高岭石外其他3种矿物与己烷之间的静电能均为正值,说明高岭石与己烷相互作用时静电能起吸附作用,其他3种矿物与己烷之间的静电能起排斥作用。甲苯与矿物表面相互作用能均大于己烷与同种矿物表面的相互作用能,说明在相同条件下,矿物表面对甲苯的吸附作用更强。
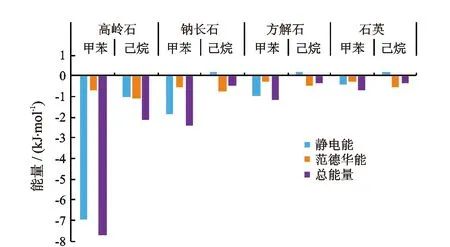
图9 甲苯和己烷与4种矿物的相互作用能
3.5 自扩散系数
油分子在4种矿物表面自扩散系数见表2。甲苯在4种矿物表面的自扩散系数均小于己烷,结合前文的相互作用能分析认为,甲苯作为一种极性分子与矿物表面的相互作用力大于己烷这种非极性分子,导致甲苯在矿物表面的吸附作用更强,更加不容易扩散。甲苯和己烷在4种矿物表面的自扩散系数的大小关系均为石英>方解石>钠长石>高岭石,与前文的相互作用能大小成反比,说明了油分子与矿物表面的相互作用力越强,油分子在矿物表面越不容易扩散。

表2 甲苯和己烷在4种矿物中的自扩散系数
3.6 径向分布函数
径向分布函数可以解释为以空间中一个粒子为中心,去寻找周围粒子的概率,即粒子在周期性边界条件下的区域密度和全局密度的比值[25]。分子动力学计算径向分布函数的方法为:
(4)
式中:g(r)为径向分布函数;ρ为空间的密度;N为粒子的数目;T为计算的时间(步数);δr为设定的距离差;ΔN为介于r→r+δr间的粒子数目。
径向分布函数的值为0时对应的最大横坐标值为粒子之间的最小间距,径向分布函数的峰值对应的横坐标值为大部分粒子之间的间距,峰值越大,表示该粒子间距对应的区域密度越大,粒子的聚集程度越高。
分子动力学平衡之后,4种矿物表面甲苯和己烷分子间的径向分布函数如图10所示。从图中可知,当分子之间的距离小于0.375 nm时,甲苯和己烷的径向分布函数都为0,这表示油分子在4种矿物表面的最近的间距不会小于0.375 nm。甲苯在钠长石和高岭石表面的径向分布函数都存在一个峰值,钠长石峰值对应的距离为0.725~0.825 nm,高岭石峰值对应的距离为0.675~0.775 nm,且钠长石的峰值大于高岭石的峰值,说明甲苯在钠长石表面分布得更集中;甲苯在方解石和石英表面的径向分布函数峰值相对不明显,表示甲苯在这2种矿物表面的分布比较分散。己烷在4种矿物表面的径向分布函数的曲线具有一致性,曲线比较平滑,没有突出的峰值,说明己烷在4种矿物表面的排列比较分散。结合前文的分析可知,矿物的亲油性越强,油分子的极性越高,油分子的排列越紧密。

图10 甲苯和己烷在4种矿物表面的径向分布函数
4 结 论
(1)通过分析甲苯和己烷在4种矿物的接触润湿角得到了4种矿物的亲油性大小关系为高岭石>钠长石>方解石>石英。
(2)矿物的亲油性越强,表面油分子的密度越大,排列越紧密,越不容易扩散,并且真空层中的油分子会加速运动到表面。
(3)矿物表面与油分子的相互作用力越强,矿物越亲油。4种矿物吸附甲苯时静电力和范德华力都起吸附作用,但以静电力为主。4种矿物吸附己烷时以范德华力为主,静电力在钠长石、方解石和石英中起排斥作用,只有在高岭石中起吸附作用。
(4)以甲苯为代表的页岩油极性物质在同种矿物表面的润湿接触角更小,吸附密度更高,排列更紧密,相互作用力更强,吸附更稳定,说明矿物对页岩油中极性物质的吸附作用强于非极性物质。
猜你喜欢
硅酸盐通报(2023年2期)2023-03-14 13:19:22
矿产综合利用(2020年1期)2020-07-24 08:51:26
中国油脂(2020年7期)2020-07-14 11:13:48
山东冶金(2018年6期)2019-01-28 08:14:42
中国粮油学报(2017年5期)2017-07-19 12:47:30
中国非金属矿工业导刊(2016年4期)2017-01-04 08:05:13
广西科技大学学报(2016年1期)2016-06-22 13:10:38
橡胶工业(2016年9期)2016-02-24 02:05:09
科技与创新(2015年20期)2015-10-29 23:36:33
超硬材料工程(2013年1期)2013-05-16 09:48:18
