
食品有害物诱发细胞焦亡的研究进展
2022-07-25 08:49李浩袁莉骆莹
食品与发酵工业 2022年13期
李浩,袁莉,骆莹
(陕西师范大学 食品工程与营养科学学院,西部果品资源高值利用教育部工程研究中心,陕西 西安,710119)
2001年,COOKSON等[1]首次使用细胞焦亡(pyroptosis)来形容在巨噬细胞中发现的半胱天冬酶-1(Caspase-1)依赖性细胞死亡方式,并证实细胞焦亡是一种新的程序性细胞死亡方式,其特征为依赖于Caspase-1活化,并伴有大量促炎症因子释放。直至2015年,邵峰院士团队研究发现,Caspase-1并不是细胞焦亡发生的必要条件,细胞焦亡还可由脂多糖(lipopolysaccharides,LPS)激活 Caspase-4/5/11引起,活化的Caspase-4/5/11通过切割激活Gasdermin家族蛋白最终诱发细胞焦亡[2-3]。因此,细胞死亡命名委员会进一步修正了细胞焦亡的定义:由Gasdermin家族蛋白介导的质膜膜孔形成的可调控性细胞死亡,经常但并不总因炎症性Caspase的活化而完成的细胞程序性坏死[4]。
1 细胞焦亡的概念及形态学特征
细胞焦亡的形态学特征、发生及调控机制等均不同于凋亡、坏死等其他细胞死亡方式(表1)[5-8]。当细胞发生焦亡时,细胞核浓缩、DNA碎片化,细胞膜上出现直径10~20 nm孔洞,细胞电解质平衡丧失,细胞肿胀破裂、胞内容物外释,激发固有免疫并使之过度活化,最终导致细胞炎性死亡。可通过透射电子显微镜观察细胞膜孔洞生成,以及Caspase-1 FLICA和SYTOX blue染色呈现双阳性来观测细胞是否发生焦亡。

表1 细胞焦亡、凋亡、自噬、坏死的特征Table 1 Characteristics of pyroptosis,apoptosis,autophagy and necrosis
2 细胞焦亡途径及相关蛋白
细胞焦亡,是一种依赖炎性Caspase蛋白,并伴有炎症反应的程序性细胞死亡方式,长期、过度的细胞焦亡会导致细胞死亡、机体组织损伤、器官衰竭、脓毒性休克等病理状态[9-10]。最新研究表明,细胞焦亡本质上由炎性Caspase蛋白切割其共同底物Gasdermin-D(GSDMD)蛋白形成的N端结构域(GSDMD-NT)介导发生,即GSDMD蛋白才是细胞焦亡的执行者[2]。
2.1 Caspase家族与NLRP3炎症小体
Caspase家族全称为半胱氨酸天冬氨酸蛋白酶,共有14个成员,在细胞死亡及炎症过程中发挥核心作用。根据细胞生物学功能和底物特异性将Caspase家族蛋白分为两大类:与炎症相关的Caspase和与细胞凋亡相关的Caspase。其中,炎症相关的Caspase也与细胞焦亡密切相关[11],包含:Caspase-l、Caspase-4、Caspase-5、Caspase-11。
炎症小体是一种多聚蛋白复合物。已发现的炎症小体有NLRP3、AIM2、Pyrin、NLRC4、NLRP1,其中NLRP3炎症小体的研究最为广泛、深入。NLRP3炎症小体由NLRP3受体蛋白先后募集凋亡相关斑点样蛋白(apoptosis associated speck-like protein containing a CARD)和Caspase-1前体(Pro-Caspase-1)构成。NLRP3炎症小体形成后,Pro-Caspase-1蛋白会自剪切形成具有活性的Caspase-1,从而启动细胞焦亡。据报道,细胞内、外源刺激物或危险信号,如:二氧化硅、石棉、细菌、病毒、真菌、细菌毒素、三磷酸腺苷等,均可激活NLRP3炎性小体,继而诱发细胞焦亡[12]。
2.2 细胞焦亡途径
最新研究指出,细胞焦亡是GSDMD蛋白介导发生的一种程序性细胞死亡方式,同时伴有大量炎性因子释放,如IL-1β和IL-18等[2-3]。它是机体受到病原微生物侵袭时启动的一种保护性免疫防御反应,但是长期、过度的细胞焦亡会诱发多种炎症性和免疫性疾病。细胞焦亡广泛参与动脉粥样硬化、肝炎和肝纤维化等疾病的发生发展[13-14]。
根据依赖的炎性Caspase和外界刺激不同,细胞焦亡被分为依赖Caspase-1的经典焦亡途径和依赖Caspase-4/5/11(Caspase-4、Caspase-5或Caspase-11)的非经典焦亡途径(图1)[15]。其中,经典焦亡途径:细胞中Caspase-1蛋白以无活性酶原形式(Pro-Caspase-1)存在。当细胞受到外界刺激(有毒物质、病原体、辐射等)后,Pro-Caspase-1通过构成NLRP3等炎症小体而被激活成具有酶活性的Caspase-1,Caspase-1继而特异性地将GSDMD蛋白切割活化形成N端结构域(GSDMD-NT)和C端结构域(GSDMD-CT),GSDMD-NT进一步通过寡聚化在细胞膜上形成膜孔,最终导致细胞破裂、死亡;同时,活性Caspase-1将促炎性因子白介素1β前体(Pro-IL-1β)和白介素18前体(Pro-IL-18)剪切为成熟型IL-1β和IL-18,经膜孔释放,并通过诱导其他炎性因子(如 IL-6,TNF-α)、黏附分子和趋化因子等合成来扩大炎症反应。非经典焦亡途径:细胞受到细菌LPS刺激后,LPS脂质部分直接与Caspase-4/5/11结合促进它们活化;活化的Caspase-4/5/11剪切GSDMD形成GSDMD-NT,启动焦亡进程;同时,GSDMD-NT也可通过激活NLRP3炎症小体活化Caspase-1,Caspase-1进而剪切Pro-IL-1β形成IL-1β,并外释、产生炎症。

图1 细胞焦亡途径Fig.1 Pyroptosis pathway
2.3 GSDMD蛋白在细胞焦亡中的作用
GSDMD蛋白是Gasdermin家族一员,是所有炎性Caspase(Caspase-1/4/5/11)的共同底物,也是人类和鼠科动物细胞焦亡的直接执行者[3]。发生细胞焦亡时,GSDMD蛋白被活化的Caspase-1/4/5/11蛋白切割成具有亲脂性和成孔活性的GSDMD-NT,以及具有亲水性和结构自抑作用的GSDMD-CT;GSDMD-NT继而选择性地与细胞膜内膜结合并寡聚化形成直径10~20 nm的膜孔,引起细胞膜渗透性紊乱,最终导致细胞溶胀、破裂,发生焦亡性死亡;同时,GSDMD-NT还能促进炎性因子IL-1β和IL-18大量释放[16-17],激发强烈的炎症反应。
3 食品有害物诱发肝脏组织细胞焦亡的相关分子机制
肝脏是食品有害物损害机体的主要靶器官。大量研究报道[23-25,28],食品有害物可通过诱导肝细胞焦亡引发一系列肝脏疾病。如苯并(α)芘、黄曲霉毒素B1、As2O3等(表2);且相关分子机制如图2所示。

图2 食品有害物诱发细胞焦亡的相关分子机制Fig.2 Molecular mechanism of cell pyroptosis induced by food hazards
3.1 苯并芘
苯并芘作为一种多环芳烃类物质,是由苯与芘稠合而成。它有2种同分异构体,分别是:苯并(α)芘(3,4-苯并芘)和苯并(e)芘(1,2-苯并芘),前者毒性更强[18]。一方面,食品在油炸、烧烤等高温条件下,其中的油脂、胆固醇等会发生热裂解反应,经环化、聚合,从而生成苯并(α)芘[19];另一方面,苯并(α)芘由人类生活、生产中使用的煤炭、石油等燃料不完全燃烧产生[20]。肝脏是苯并(α)芘的主要靶器官[21-22]。前期研究发现,20 μmol/L苯并(α)芘处理HL-7702人肝细胞24 h后,细胞相对电导率、NO释放量、乳酸脱氢酶(lactate dehydrogenase,LDH)释放率显著升高;同时HL-7702人肝细胞内焦亡特征指标Caspase-1、IL-1β、IL-18的蛋白表达量增多[23]。说明苯并(α)芘可诱导HL-7702人肝细胞发生焦亡性损伤。基于此,LI等[24]研究发现天然黄酮异荭草素通过降低电导率、LDH和NO水平,抑制Pro-Caspase-1、Cleaved-Caspase-1、iNOS、Cox-2蛋白表达量,降低IL-1β和IL-18的mRNA水平,减弱由苯并(α)芘诱导的人肝细胞焦亡性损伤。同时,有研究证明苯并(α)芘通过PI3K/Akt信号通路诱导HL-7702人肝细胞焦亡性死亡[25]。
3.2 黄曲霉毒素B1
黄曲霉毒素是一组毒性极强的二吹喃香豆素的衍生化合物,1993年被国际癌症研究机构(International Agency for Research on Cancer,IARC)认定为I类致癌物[26]。其中,黄曲霉毒素B1毒性最强,可严重损伤肝脏组织[27]。张力引[28]研究发现,采用1 mg/kg黄曲霉毒素B1隔天灌胃C57BL/6小鼠,4周后处理组小鼠肝组织中NLRP3、ASC、IL-1β、GSDMD的蛋白水平升高、IL-1β分泌增多,表明黄曲霉毒素B1能诱发肝脏组织炎症反应;另外采用20 μmol/L黄曲霉毒素B1对HepaRG人肝细胞进行处理,24 h后处理组细胞LDH释放率、IL-1β、活性氧(reactive oxygen species,ROS)含量均显著升高,NLRP3、ASC、IL-1β、GSDMD蛋白表达量增多,焦亡细胞数目增多。张力引[28]在成功建立焦亡细胞模型的基础上利用环氧合酶-2去磷酸化修饰调控处理组细胞,结果显示焦亡细胞数目、NLRP3等焦亡相关蛋白表达量有所降低,同时间接说明黄曲霉毒素B1可诱导肝细胞发生依赖于NLRP3炎症小体的焦亡性损伤。
3.3 As2O3
大量流行病学调查显示,砷的长期暴露会导致肝损伤、心血管代谢异常等疾病[29]。其中,肝脏是砷代谢的主要器官[30]。邱天明[31]研究了As2O3诱导非酒精性脂肪肝/非酒精性脂肪性肝炎的相关机制,结果发现As2O3能够促进NLRP3炎症小体、Caspase-1等参与HepG2细胞焦亡,为砷中毒相关疾病控制提供新的思路。
除以上食品有害物外,有研究发现摄入过量的铜离子和长期高脂饮食也会导致肝脏组织细胞焦亡性损伤。SU等[32]发现鸡肝细胞经CuCl2处理后ROS含量、膜电位升高,焦亡细胞数目增多,说明Cu2+通过刺激肉鸡肝线粒体呼吸产生大量ROS,造成细胞脂质过氧化,发生焦亡性损伤。张裕恒[33]研究结果表明,活性氧清除剂NAC、Caspase-1抑制剂Z-YVAD-FMK的加入均可降低鸡肝细胞中Caspase-1、IL-1β、IL-18的蛋白表达量,表明Cu2+是通过ROS介导的Caspase-1途径诱导鸡肝细胞焦亡。此外,随着人们生活水平不断提高,高脂肪食物摄入量占饮食结构的比例逐年升高。大量研究表明,高脂饮食是肝脏疾病发生发展的主要诱因之一[34]。陈璐等[35]研究发现C57BL/6小鼠摄入高脂饮食后,肝组织中Caspase-1、NLRP3等蛋白表达量升高,同时使得下游炎症因子IL-1β分泌变多,从而导致焦亡细胞增多,肝脏损伤加重,充分证实NLRP3-Caspase-1信号通路与焦亡的关联性。
唐标等[36]指出,降脂理肝汤能够减轻高脂饮食诱导的非酒精性脂肪肝症状,其干预机制与抑制非经典的细胞焦亡途径有关。降脂理肝汤的干预能显著降低Caspase-11的水平,较低的Caspase-11水平会影响GSDMD蛋白活化,进而减轻非酒精性脂肪肝症状。同时,吴柳等[37]利用下瘀血汤干预改善非酒精性脂肪性肝炎,其机理之一是降低NLRP3蛋白表达、下调NLRP3炎症小体相关基因。因此可将细胞焦亡关键基因作为靶点,研发干预和治疗非酒精性脂肪肝/非酒精性脂肪性肝炎的相关保健食品和药品。
4 食品有害物诱发免疫组织细胞焦亡的相关分子机制
免疫细胞是免疫系统的主要组成部分,在防御病原体入侵、协调机体系统等方面有重要作用。当机体细胞因外来病原微生物入侵可能发生焦亡性死亡时,免疫细胞将会被招募用于清除病原物,以维持机体内环境稳定,从而缓解机体细胞的焦亡性损伤[38]。据报道,一些食品有害物也可通过诱发免疫组织细胞焦亡损害机体健康,如表2所示。
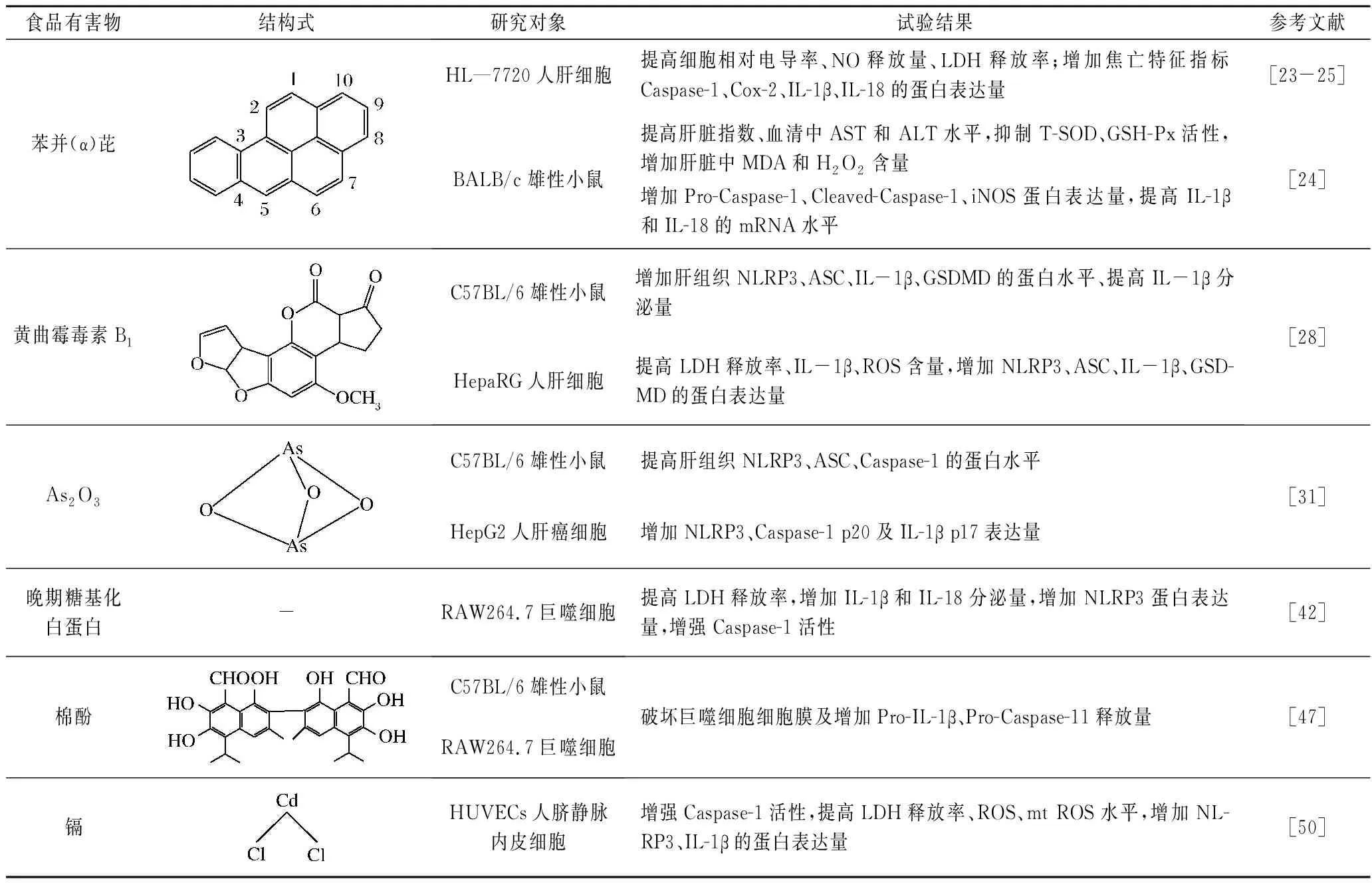
表2 食品有害物诱发细胞焦亡性损伤Table 2 Food hazards induce pyroptosis
4.1 晚期糖基化产物
晚期糖基化终产物(advanced glycation end products,AGEs)是葡萄糖或其他还原糖(如半乳糖和果糖)与氨基酸、核苷酸碱基或脂肪酸反应形成糖基化分子时形成的一组不同的化合物[39]。体内AGEs水平过高可导致疾病[40]。大量研究表明,AGEs通过与细胞表面受体结合或与机体蛋白质交联,改变其结构和功能,从而促进氧化应激和炎症反应[41]。
晚期糖基化白蛋白(advanced glycated albumin,AGE-alb)是AGEs的代表物质之一。张昭强等[42]使用AGE-alb预处理RAW264.7巨噬细胞,与空白组相比细胞活力降低,LDH释放率升高、IL-1β和IL-18分泌增多,NLRP3蛋白表达量增加,Caspase-1活性增强,充分证明AGE-alb成功诱导RAW264.7巨噬细胞焦亡性损伤。为验证AGE-alb是否通过NLRP3-Caspase-1信号通路引起细胞焦亡,张昭强等[42]采用NLRP3特异性抑制剂MCC950作用RAW264.7巨噬细胞,结果发现MCC950可以显著抑制AGE-alb诱导的细胞焦亡,同时证实AGE-alb通过NLRP3-Caspase-1信号通路诱导免疫细胞焦亡性损伤。
4.2 棉酚
棉酚(gossypol,GOS)是从棉花的种子、根和茎中提取的天然产物[43]。GOS一方面因为多种药理学活性,在防御害虫和病原体方面发挥重要作用[44]。另一方面因其细胞毒性,可诱导山羊精原干细胞、小鼠生精细胞发生细胞凋亡[45-46]。GOS作为一种具有毒性的多酚类化合物,关于其是否对免疫细胞中炎症小体和细胞焦亡产生影响等,目前尚未清楚。为此,林秋茹[47]以小鼠腹腔巨噬细胞以及RAW 264.7细胞系为研究对象,进行深入探究。结果发现GOS可以诱导经LPS刺激产生的巨噬细胞细胞膜破碎及Pro-IL-1β、Pro-Caspase-11的大量释放,最终使其发生细胞焦亡;GOS也可以诱导缺乏ASC的RAW 264.7细胞发生焦亡,说明GOS诱导巨噬细胞焦亡并不依赖于经典的Caspase-1活化。虽然GOS可以致使大量Pro-IL-1β的产生,但仅能诱导低水平的成熟IL-1β的分泌。同时,利用其他的经典焦亡抑制剂并没有能够抑制GOS诱导的细胞焦亡,表明GOS可能是通过非经典炎症小体通路诱导细胞发生焦亡性损伤。
4.3 镉
大量研究显示,摄入过量的镉会导致免疫组织损伤,引发骨质疏松症、肾功能不全、糖尿病、癌症等[48]疾病。血管内皮细胞是镉的重要靶细胞,镉会通过诱导血管内皮细胞释放多种炎性因子,产生一系列心血管疾病[49]。为研究镉诱导细胞焦亡的机制,陈海燕[50]使用80 μmol/L CdCl2对HUVECs人脐静脉内皮细胞进行处理,24 h后镉处理组HUVECs人脐静脉内皮细胞Caspase-1活性增强,LDH释放率升高,细胞发生焦亡性损伤。另外分别使用Caspase-1抑制剂Z-YVAD-FMK、NLRP3 siRNA以及ROS和mt ROS特异性抑制剂NAC和Mito-TEMPO对HUVECs细胞进行预处理,结果显示均可以缓解细胞发生焦亡性损伤。因此镉诱导细胞发生焦亡的机制为:首先通过诱导氧化应激,产生大量ROS和mt ROS刺激活化NLRP3炎症小体,从而发生Caspase-1的经典焦亡途径。
5 小结
细胞焦亡是一种不同于坏死、自噬、凋亡等的细胞死亡方式,它分为依赖Caspase-1的经典焦亡途径和依赖Caspase-4/5/11的非经典焦亡途径。食品有害物可通过诱导肝脏组织细胞和免疫组织细胞发生焦亡,损伤机体健康。全面深入研究细胞焦亡的分子机制,可以为食品有害物毒性控制提供新的思路。目前研究发现,细胞焦亡的发生会引发各种炎症反应,因此应当更加着眼于充分了解焦亡参与炎症反应发生发展的关系和机制,将有助于开发相关靶向药物,对临床患者的康复治疗具有重要意义。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
世界科学技术-中医药现代化(2022年3期)2022-08-22
材料与冶金学报(2022年2期)2022-08-10
医学综述(2022年7期)2022-04-19
昆明医科大学学报(2021年12期)2021-12-30
科学与财富(2021年33期)2021-05-10
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年9期)2021-01-18
作文成功之路·小学版(2020年6期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
