
基于微生物16SrRNA测序技术对ICR小鼠传代后肠道菌群变化的分析
2022-07-25 06:42:06张璐瑶颛清芮傅祥伟侯云鹏
中国农业大学学报 2022年7期
张璐瑶 孟 琳 颛清芮 傅祥伟,3 侯云鹏*
(1.中国农业大学 生物学院/农业生物技术国家重点实验室,北京 100193;2.中国农业大学 动物科技学院/畜禽育种国家工程实验室,北京 100193;3.新疆农垦科学院 省部共建绵羊遗传改良与健康养殖国家重点实验室,新疆 石河子 832000)
哺乳动物胃肠道内定植了1 014种细菌,这些细菌基因总和是宿主基因数目的 100 多倍,因此也被称为寄主体内的“隐形器官”。近年来关于哺乳动物肠道微生物的研究表明,肠道微生物可以通过调节内分泌和神经等方式参与机体的能量代谢、物质吸收、免疫调节等多项生命活动。作为宿主体内控制代谢的隐形器官,肠道微生物失调可能造成多种代谢紊乱疾病,如肥胖,Ⅱ型糖尿病,脂肪肝和动脉粥样硬化等。此外,肠道菌群易受宿主年龄、性别以及饮食等应激性环境因素的影响。
小鼠肠道中存在大量的微生物菌群,这些微生物菌群结构复杂种类繁多,参与宿主的代谢,在宿主的消化吸收和免疫调控方面均发挥了重要作用,对小鼠的生长发育有很大影响。ICR品系小鼠以多产为目标进行选育,由美国癌症研究所(Institute of cancer research)分送各国饲养的实验小鼠ICR。ICR品系小鼠毛色白化,同时具有适应性强,体格健壮,繁殖力强,生长速度快,实验重复性好等特点,是常见的实验室模式动物。结合本实验室和我国的从事实验动物饲养管理的研究者发现,经过长时间和多代次的近亲扩繁,ICR品系小鼠随着繁殖代次的增加会出现一些生长和繁殖性能异常的状况。经统计分析后发现,ICR品系小鼠传代后新生子代的生长速度显著降低,各年龄段体重显著降低,体长显著降低等现象。
随着基因组技术和二代测序技术的发展,研究微生物群落结构也实现了从传统的细菌培养基提取法到使用高通量测序技术的转变。其中,16SrRNA测序已经成为研究微生物群落组成及其分布的重要手段。16SrRNA测序技术是通过比对细菌 16SrRNA 序列对细菌进行快速种属鉴定的一种方法。与此同时16SrRNA测序技术还可以对样品中的优势物种以及一些未知的物种进行检测和分类,获得样品中的微生物组成并计算出它们的相对丰度。此外,16SrRNA测序还可以通过差异菌群进行差异代谢通路预测分析,预测肠道微生物的变化带来的代谢差异表型等。由此,猜测ICR品系雄性小鼠随着繁殖代次的增加出现的生长异常可能与其肠道微生物的改变有关。为探究ICR雄性小鼠随着繁殖代次的增加出现的生长异常和其肠道微生物的关系,分别收集F0、F1、F2和F3代18周雄鼠的大肠内容物,每组收集6个重复,通过16SrRNA测序技术,研究ICR雄性小鼠传代之后肠道菌群的变化。
1 材料与方法
1.1 试验材料
1
.1
.1
试验动物清洁级雌性ICR小鼠20只,雄性ICR小鼠10只(北京维通利华实验动物技术有限公司)7周龄,体重25~30 g。实验动物饲养于中国农业大学西校区动物实验室,小鼠繁殖饲料喂养,自由进食、饮水。小鼠繁殖饲料具体营养成分见(表1),从华阜康实验动物公司购买。动物实验室严格按照相关参数进行控制:小鼠自由采食和饮水,供应充足,垫料每周更换一次。光照控制在12 h∶12 h(北京时间8:00~20:00光照),温度控制在20~25 ℃,湿度为45%~55%。

表1 饲料配方Table 1 Feed formula
1.2 试验方法
1
.2
.1
动物建模方式所有小鼠均在清洁级鼠房饲养一周以适应环境,将这些小鼠作为F0代记录。随后,标记耳号(耳号按繁殖代次标记为:F0:10XX)并记录后,每笼按照雌雄比2∶1进行合笼。雌鼠明显怀孕后,取出单笼饲养。产仔记为F1代,及时记录产仔数与出生体重(注意初产雌鼠易食仔,需及时查看处理;若雌鼠流产导致妊娠失败也需记录)。幼鼠F1代21日龄时断奶,标记耳号并记录性别与离乳体重,将饲料剥碎放入幼鼠笼内便其采食。35日龄(性成熟)时,取体况良好的F1代雄鼠与雌鼠1∶2交配进行下一代繁殖实验,此时应注意避免近亲交配,产仔标记为F2代。处理过程与上述F0代时处理相同。以此类推,F2代雄鼠和雌鼠1∶2交配后产生的后代记为F3代,以此类推,将其后代分别标记为F1:11XX,F2:12XX,F3:13XX,F4:14XX, F5:15XX。
1
.2
.2
肠道微生物样本收集小鼠处死后,取大肠中的内容物于无菌冻存管中,分别收集F0、F1、F2和 F3 代 18周 雄鼠的大肠内容物,每组收集6个重复。标记好后立即投入液氮中迅速冷冻。待所有样本收集完成后,转移至-80 ℃超低温冰箱中保存。
1
.2
.3
肠道微生物样本DNA提取大肠内容物标本解冻以后,按照Fast DNA SPIN Soil Kit (MP Biomedicals,Santa Ana,CA)说明书的操作说明进行样本DNA提取,提取细菌DNA。所有样品全部提取完成后,放入干冰中送检,使用Qiime平台进行16SrRNA测序。后续的细菌DNA扩增和数据分析均由北京奥维森科技有限公司承担。
1
.2
.4
细菌DNA扩增对肠道微生物样本的16SrRNA V3~V4可变区进行PCR扩增,引物为(F:ACTCCTACGGGAGGCAGCAG R:GGACTACHVGGGTWTCTAAT 北京奥维森科技有限公司)。选择Premix Taq 试剂。PCR扩增反应体系如下:94 ℃预变性5 min; 94 ℃ 30 s,55 ℃ 30 s,72 ℃ 1 min, 28个循环; 72 ℃延伸7 min。然后使用1%琼脂糖凝胶电泳对样本质量进行定量检测,最终选择DNA浓度>50 mg/L、总量>3 ng的24份样品进行16SrRNA测序。
1
.2
.5
细菌16SrRNA测序分析使用16SrRNA的高可变区QIIME测序技术,对样品的16SrRNA V3区PCR产物进行测序,结果以“Fast-q”格式存储。同时,使用Mi-seq测序得到Pair-End(PE)双端序列数据,对测得的数据进行质控分析,经过一系列的筛选,拼接和去冗余处理等,最终得到优质的数据。质控分析过程中,需要使用Trimmomatic (V0.36),Pear (V0.96),Flash (V1.20),Vsearch (V2.7.1)等相关软件。
将序列按照相对丰度排序,排列顺序从高到低。所有序列依据不同的相似度进行OTU划分,对大于97%相似水平的OTU进行生物信息分析。结合QIIME(version 1.9.1)平台,参考16S细菌和古菌核糖体数据库, kSilva2(Release128/132 http:∥www.arbsilva.de),RDP(V16 http:∥rdp.cme.msu.edu/),Greengene(Release13.5 http:∥greengenes.secondgenome.com/)采用RDP classifier贝叶斯算法进行分类学分析。最后通过序列比对获得每个OTUs的物种分类信息。使用PICRUSt对OTUs丰度标准化,并将每个标准化的OTUs对应到相应的基因进化树,从而得到相应的KEGG信息,并用STAM P对得到的KEGG进行差异分析。
1
.2
.6
统计分析使用SPSS分析软件中的独立样本t
检验对小鼠的体重,微生物多样性以及特定菌种差异分析。P
<0.05为显著性差异,P
<0.01为极显著性差异,P
>0.05为差异不显著。2 结果与分析
2.1 不同繁殖代次ICR雄性小鼠在相同周龄的体重分析
为了探究不同代次ICR品系小鼠的生长发育情况,在0、3、5和18周4个时期分别取F1、F2、F3、F4和F5 代雄性小鼠进行称重,每组样本数不低于20只。统计结果如图1所示,在0、3、5和18周,小鼠的体重随着代次的积累均呈下降趋势,且差异显著,说明ICR雄性小鼠随着繁殖代次的增加,体重显著降低。

*P<0.05,**P<0.01,***P<0.001,ns差异不显著(P>0.05)。下同。* Represents P<0.05, ** Represents P<0.01, *** Represents P<0.001, ns represents significant difference at 0.05 level。The same below.图1 不同代次ICR雄性小鼠在相同周龄的体重比较Fig.1 Comparison of body weight of ICR male mice of different generations at the same age
2.2 不同繁殖代次ICR雄性小鼠肠道微生物测序分析
为探究ICR雄性小鼠随着繁殖代次的增加出现的生长发育异常和其肠道菌群关系,分别收集F0、F1、F2和 F3 代 18周雄鼠的大肠内容物,每组收集6个重复,对16S的V3~V4可变区进行16SrRNA测序。根据16SrRNA的测序结果,采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,参考16 s细菌和古菌核糖体数据库Silva,共产生 1 786 个 OTU,经过抽平处理剩余1 729个。如图2(a)示,F0代有OTUs 有1 276个,其中特殊OTUs 有87个;F1代有OTUs 有1 258个,其中特殊OTUs 有113个;F2代有OTUs 有1 173个,其中特殊OTUs 有47个;F3代有OTUs 有1 195个,其中特殊OTUs 有47个。为了更好的显示多个样本之间的距离关系,将加权与未加权的unifrac样本距离矩阵用热力图表现出来,如图2(b)所示,聚类结果表明F0、F1、F2和 F3 代各组间微生物群落物种有明显差异。
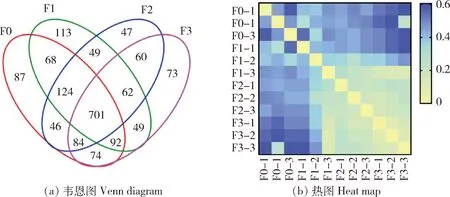
图2 不同代次ICR雄性小鼠肠道微生物测序分析Fig.2 Intestinal microbial sequencing analysis of different generations of male ICR mice
2.3 不同繁殖代次ICR雄性小鼠肠道微生物的Alpha多样性指数分析
通过对每个样品OTU总数和每个OTU的相对丰度进行样品多样性指数的计算。不同繁殖代次ICR雄性小鼠肠道微生物的Alpha多样性比较结果见图3。在分析Alpha 多样性的过程中,通常使用Shannon-Wiener指数来反映样本中微生物多样性,Shannon指数利用各样本在不同测序深度时的微生物多样性指数构建曲线,反映各样本在不同测序数量时的微生物多样性。如图3(b)所示,随样本序列数的增加,曲线逐渐趋于平坦,说明测序数据量足够反映样本中绝大多数微生物的信息,测序分析预测结果可信度较高。Chao1和Observed-species指数根据所测得OTUs的数量来预测样品中微生物种类,两者值越大,物种越丰富;由图3(c)可知,F0、F1、F2和F3的chao1指数分别为922.035±78.932 8、821.1 625±72.941 8、757.502 5±19.1 453和613.325±106.878 7,各组两两比较均有显著差异(P
<0.05),随繁殖代次的增加,chao1指数显著下降。由图3(d)可知,F0、F1、F2和F3的Observed-species指数分别为654.2±53.52 23、557.675±49.626 3、548.7±18.476 7和384.2±54.112 9,F0、F1和F2间两两比较无显著差异(P
>0.05),但F3代较前三组有显著下降(P
<0.001)。辛普森指数 (Simpson) 和香侬指数(Shannon)的计算是基于样品的所有OTU丰度和均匀度及其注释物种信息进行的。辛普森指数和香侬指数可以反映样本的菌群种类数和每个菌种占总体群落的比例, Simpson指数越接近于1,香侬指数(Shannon)指数越大,说明样品中的菌群种类越丰富,多样性水平越高。由图3(e)可知,F0、F1、F2和F3的香侬指数(Shannon)分别为5.3 275±0.607 0、5.7 375±0.481 5、4.425±0.484 1和3.5 775±0.417 7,F0和F1比较无显著差异(P
>0.05),但随代次的增加,F2和F3的香侬指数(Shannon)指数显著下降(P
>0.05)。由图3(f)可知,F0、F1、F2和F3的Simpson指数分别为0.865±0.075 50、0.902 5±0.069 56、0.877 5±0.044 25和0.815±0.03 697,各组两两比较均无显著差异(P
>0.05)。综上,结果表明随繁殖代次的增加,肠道菌群Alpha多样性呈显著下降趋势。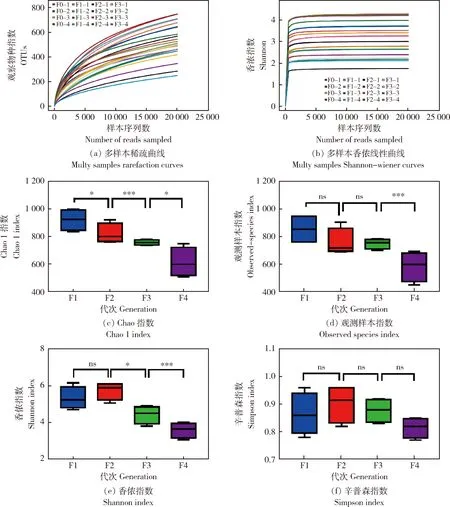
图3 不同繁殖代次ICR雄性小鼠肠道微生物的Alpha多样性比较Fig.3 Comparison of Alpha diversity of intestinal microbial in different generations of male ICR mice
2.4 不同繁殖代次ICR雄性小鼠肠道微生物群中门水平微生物类群的差异
不同繁殖代次ICR雄性小鼠肠道微生物群门水平聚类结果表明各组间微生物群落物种有明显差异。如图4(a),在门水平上,各组的优势菌群均为厚壁菌门(Firmicutes)(F0、F1、F2和F3组分别为83.3%,79.0%,81.8%和90.3%)和拟杆菌门(Bacteroides)(F0、F1、F2和F3组分别为6.0%、5.1%、4.0%和3.2%)。在F0组中拟杆菌门(Bacteroides)的丰度为6.0%,厚壁菌门的丰度为83.3%,且在F1组中拟杆菌门(Bacteroides)的丰度显著低于F0组(P
<0.01),F2和F3组也显著低于F0和F1组(P
<0.01)(图4(b))。此外,如图4(c),脱铁杆菌门(Deferribacteres)在F2和F3组中的丰度也有显著降低(P
<0.01)。综上,随代次积累,ICR品系小鼠肠道微生物多样性在门水平上有很大变化,并且随代次积累,对一些特定的菌群,如厚壁菌门、拟杆菌门和脱铁杆菌门丰度的影响更为深远。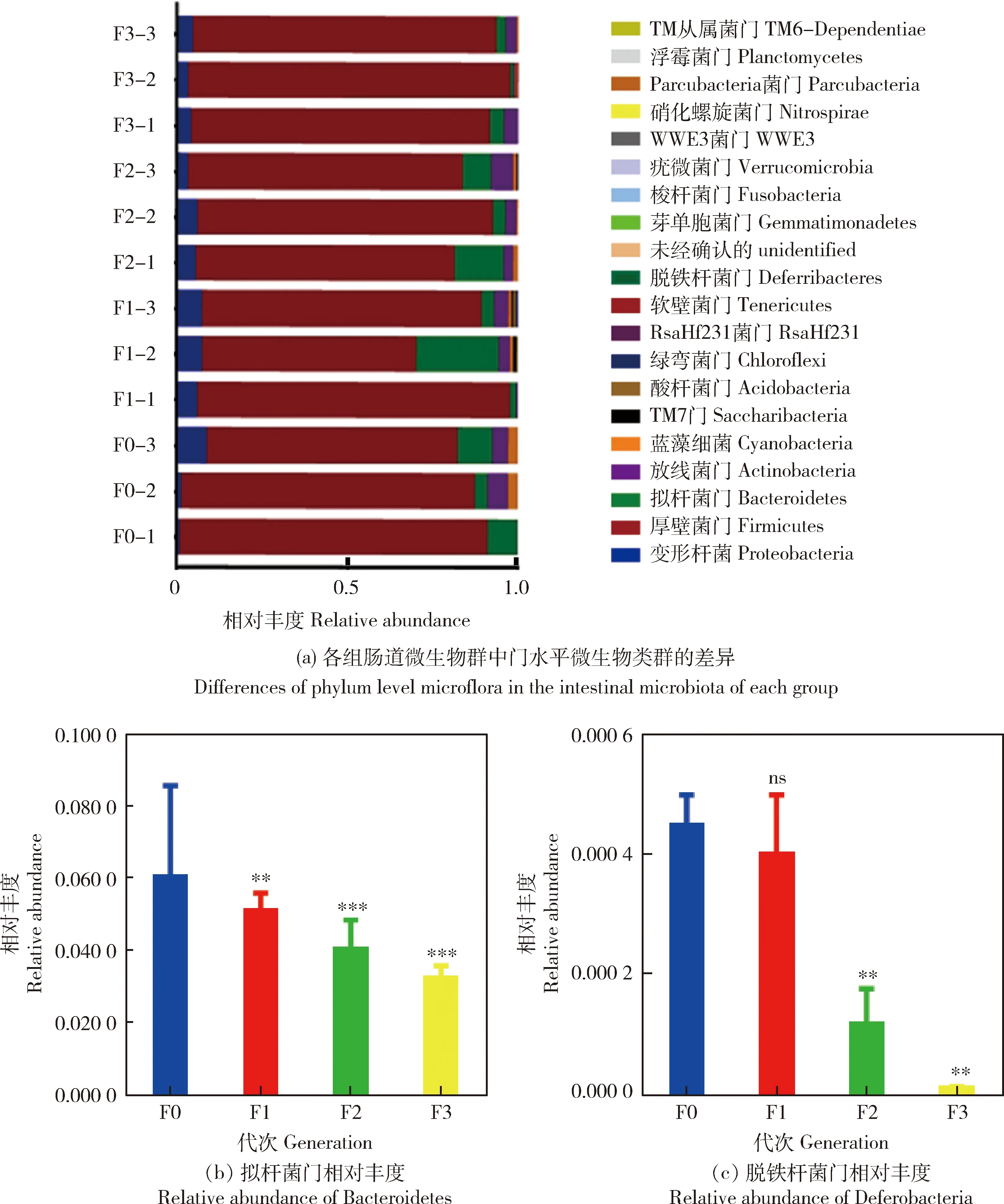
图4 不同繁殖代次ICR雄性小鼠肠道微生物群中门水平微生物类群的差异Fig.4 Differences of phylum level microflora in the intestinal microbiota of different generations of male ICR mice
2.5 不同繁殖代次ICR雄性小鼠肠道微生物群中属水平微生物类群的差异
如图5(a)所示,毛螺菌科-NK4A136组,螺杆菌属、乳酸球菌、梭菌属-sencu-stricto-1、瘤胃球菌属、弯曲杆菌属、未确认的-毛螺菌科、链球菌属、乳酸菌属、球菌属、埃希氏菌属、普氏菌属和拟杆菌-S24-7组等在各组肠道微生物群中属水平微生物类群均有差异。在属水平上,F0、F1和F2 3组的优势菌群均为乳酸菌属(Lactobacillus
)(F0、F1和F2组分别为83.1%、62.8%和38.2%),但F3代小鼠的一些未确认分类的菌属占比较高,约为45.3%。此外,如图5(b)所示,虽然F0、F1和F2三组中乳酸菌属均为优势菌群,但各组的乳酸菌属丰度有显著差异(P
<0.05),且随代次的积累呈逐渐下降的趋势。埃希氏菌属(Escherichia
)(F0、F1、F2和F3组分别为0.015%、0.032%、0.439%和2.41%)在F1、F2和F3代中的丰度有显著升高(P
<0.01)(图5c)。综上,随代次积累,ICR品系小鼠肠道微生物多样性在属水平上有很大变化,并且随代次积累,产生的影响更为深远。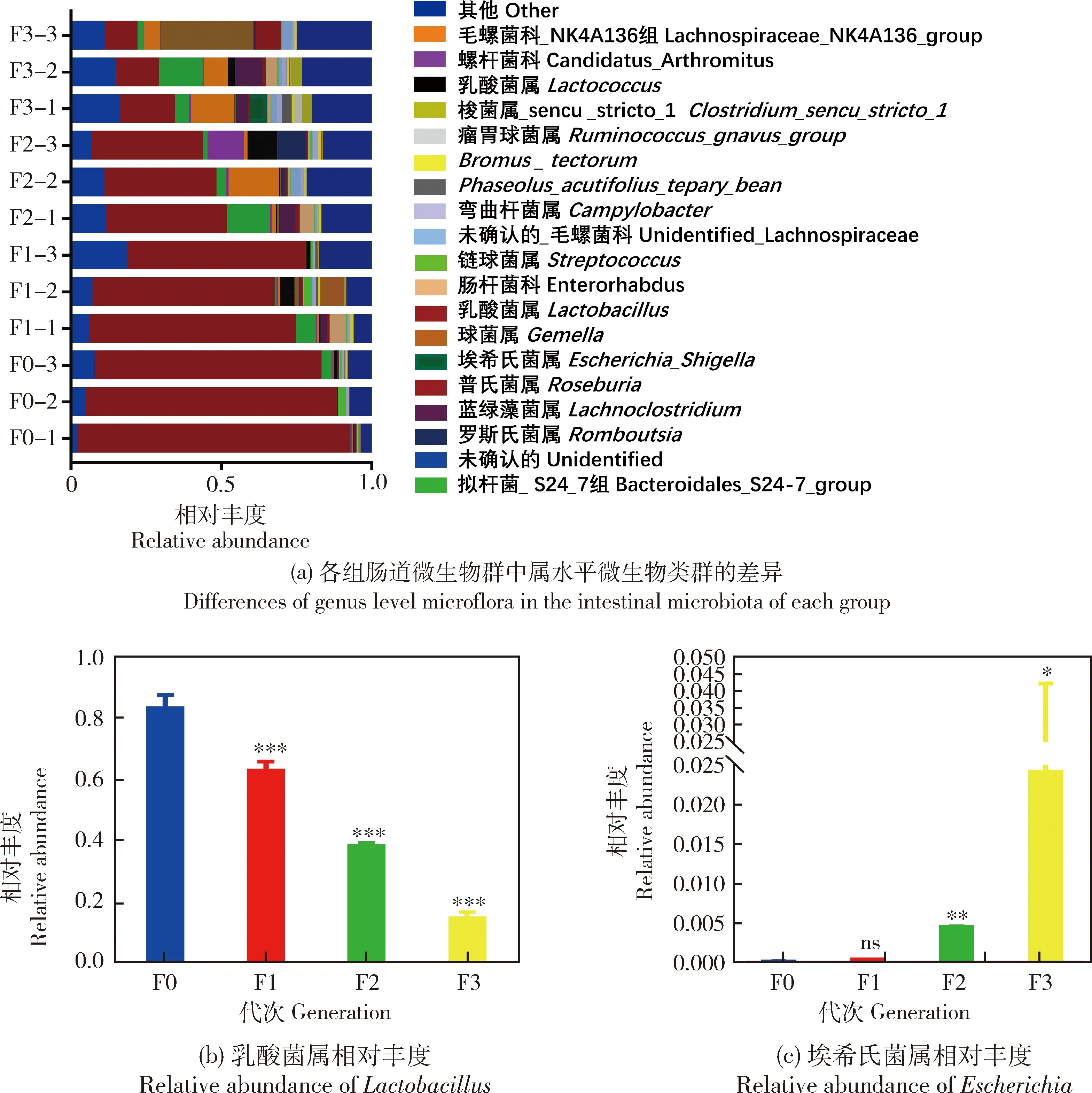
图5 不同繁殖代次ICR雄性小鼠肠道微生物群中属水平微生物类群的差异Fig.5 Differences of genus level microflora in the intestinal microbiota of different generations of male ICR mice
2.6 不同繁殖代次ICR雄性小鼠肠道微生物的差异代谢途径(KEGG)预测
为进一步探究ICR子代体重下降的机制,本研究针对这一变化出现的差异最明显的两个组,即F0代和F3代的肠道微生物进行KEGG差异代谢通路预测。如图6可知,KEGG分析发现14条代谢途径有显著差异,其中,乙醛酸盐代谢、胰岛素信号通路、苯丙氨酸,色氨酸和酪氨酸生物合成途径,萜类化合物生物合成途径,肽聚糖生物合成和糖酵解糖异生作用在F3组中的丰度显著低于F0组(P
<0.05),结果显示F3代氨基酸合成,糖类代谢等多条基础代谢通路受损。因此,不同繁殖代次ICR雄性小鼠肠道微生物的变化可能对小鼠的新陈代谢有一定影响。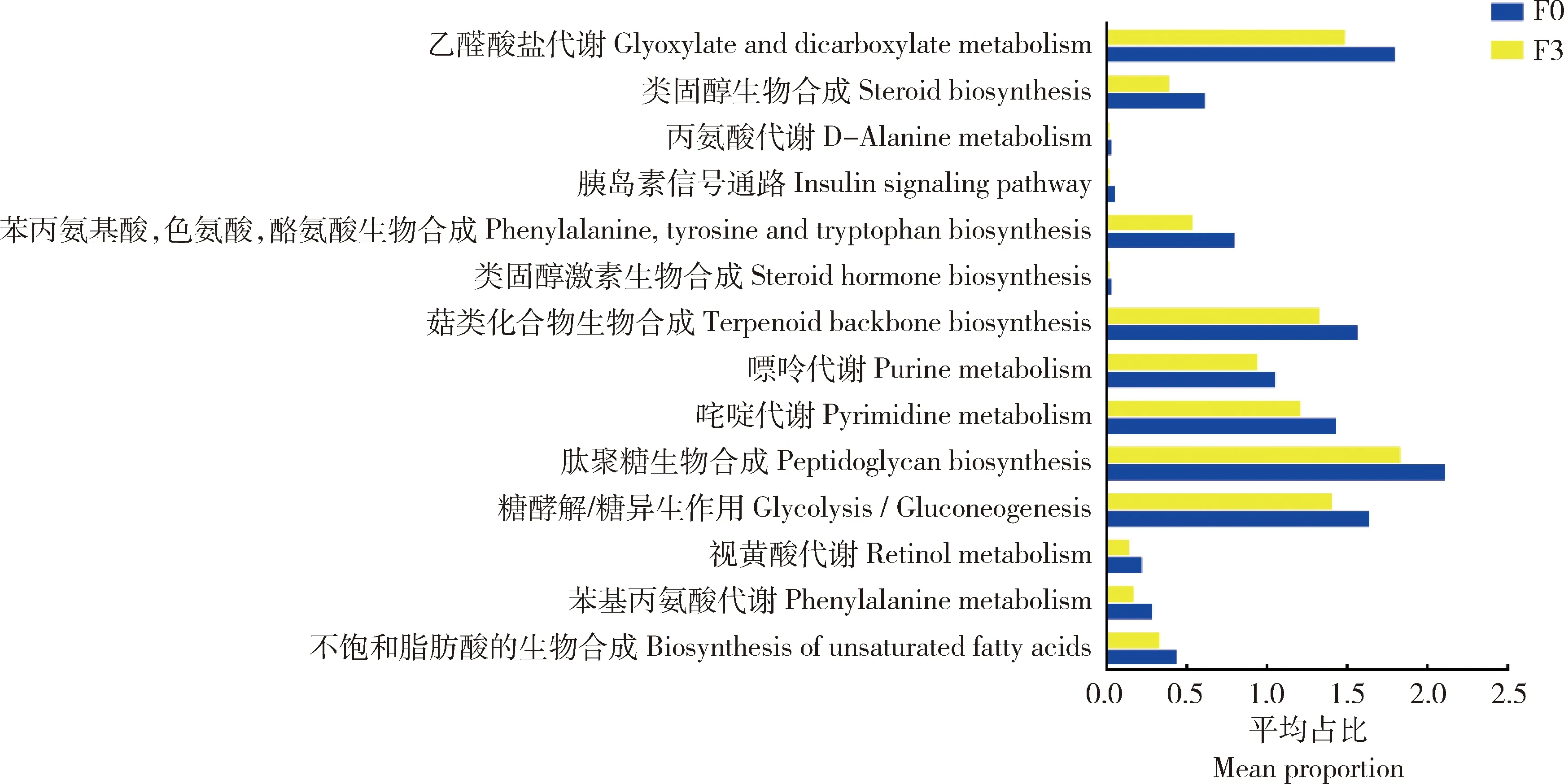
图6 F0和F3组肠道微生物差异生物功能代谢通路预测Fig.6 Analysis of metabolic pathway of biological different functions of the fecal microbiota in different generations of male ICR mice
3 讨 论
3.1 ICR雄性小鼠传代后肠道微生物多样性显著降低
本研究采用16SrRNA基因高通量测序技术分析ICR雄性小鼠随着繁殖代次增加出现的生长异常和其肠道菌群的关系。通过对测序结果的深度挖掘和分析,发现随代次积累,ICR雄性小鼠肠道微生物多样性显著降低,优势菌门如拟杆菌门丰度显著降低,相关益生菌门如脱铁杆菌门的丰度也显著降低,且随代次的积累逐渐下降。以上结果显示肠道菌群的失调可能是导致ICR雄性小鼠出现生长异常的原因。大多数细菌可被可归为两大类,一大类是厚壁菌门,包括葡萄球菌、链球菌等;另一大类是拟杆菌门。有研究表明肠道菌群多样性会影响宿主的体重,在对各组肠道菌群的分析中,不难发现拟杆菌门丰度显著降低,这些变化可能与ICR雄性小鼠体重降低有关。另外值得注意的是,在针对属水平的肠道微生物丰度分析中,发现随ICR雄性小鼠繁殖代次的积累,乳酸菌属丰度显著降低,且随代次的积累呈逐渐下降的趋势。
乳酸菌(Lactic
acid
bacteria
,LAB)是一类在动物胃肠道中广泛存在的厌氧或兼性厌氧性细菌,且此类细菌利用可发酵碳水化合物产生大量乳酸等有机酸,是一类广谱益生菌。作为益生菌,乳酸菌具有丰富的物种多样性,包含18个属,共200多种。乳酸菌在代谢过程中可以为宿主供给必需营养物质,增强代谢,促进其生长。乳酸菌还可以调节肠道环境酸碱平衡,使其符合淀粉酶和糖化酶等消化酶的最适pH,促进营养物质的消化吸收。此外,乳酸菌可以改善肠道微环境,抑制一些有害的致病菌的生长,增强生物体对疾病的抵抗力。基于此,本研究认为乳酸菌丰度的降低是造成 ICR雄性小鼠子代出现生长发育异常的一个重要原因。3.2 ICR雄性小鼠KEGG差异功能预测
除了益生菌缺失对肠道菌群多样性造成的影响之外,肠道菌群还在机体不同的代谢途径中发挥作用。差异菌群导致的代谢异常可以进一步解释ICR雄性小鼠子代出现生长发育异常表型。结合KEGG分析表明,随繁殖代次增加,ICR雄性小鼠的苯丙氨酸,色氨酸和酪氨酸生物合成等氨基酸合成途径,萜类化合物生物合成途径,胰岛素信号通路,糖酵解糖异生作用等糖类代谢途径可能会出现表达异常的情况。芳香族氨基酸中的苯丙氨酸、色氨酸属于必需氨基酸,而酪氨酸是半必需氨基酸。其中苯丙氨酸可以氧化成酪氨酸,并与酪氨酸一起合成重要的神经递质和激素,参与机体糖代谢和脂肪代谢,因此芳香族氨基酸代谢对机体的其他两大代谢途径有很重要的调节作用,芳香族氨基酸代谢失调将直接导致机体的代谢功能紊乱。此外,胰岛素信号通路在机体代谢的调控方面也有举足轻重的地位。胰岛素是能量代谢的主要调控激素,胰岛素与受体结合后,激活受体中的蛋白酪氨酸激酶(PI3K),被激活的酪氨酸激酶作用于受体底物,使受体底物发生磷酸化,随后激活3种已知的 AKT 异构体,最终通过多种不同的途径参与到胰岛素的代谢调节中,促进机体对能量利用。通过图6可以推测ICR雄性小鼠能量利用率也在逐渐降低。
3.3 展望
本研究结果初步探明ICR雄性小鼠的肠道菌群失衡可能是导致其生长发育异常的机制之一。由于机体肠道微生物的组成结构复杂,本研究仅在门和属水平对肠道菌群进行了研究,并根据KEGG分析了可能影响的基础代谢通路,对于具体关键微生物和生长发育之间的因果关系值得进一步研究。在后续的研究中,可以考虑对ICR小鼠进行乳酸菌等益生菌的灌胃试验,观察是否可以改善ICR雄性小鼠后代发育异常和体重下降等异常情况。
本研究结果同时表明,F2代前优势菌群未发生明显改变,建议使用ICR子代进行试验研究最好在F2代前,如确需更多子代进行研究,建议考虑以非近亲繁殖方式的进行交配以进行扩繁和传代。对个体一致性和遗传表型稳定性一要求较高的生物学试验ICR鼠不建议作为首选,如果实验室经费和资源有限,在使用ICR小鼠进行繁殖实验时,须定期引入亲缘关系较远的ICR小鼠进行扩繁和传代。
4 结 论
ICR雄性小鼠传代后肠道微生物多样性显著降低,拟杆菌门和脱铁杆菌门丰度显著降低,乳酸菌属丰度显著降低,埃希氏菌属等致病菌丰度显著提高。结合差异代谢通路预测分析,随代次积累ICR雄性小鼠氨基酸合成,糖类代谢等多条基础代谢通路可能受损,这些变化均可导致ICR雄性小鼠后代发育异常和体重下降等缺陷。
猜你喜欢
垂钓(2023年11期)2024-01-21 16:07:04
垂钓(2023年9期)2023-12-10 19:39:30
中老年保健(2022年2期)2022-08-24 03:20:50
现代畜牧科技(2021年9期)2021-10-13 06:38:44
科学(2020年4期)2020-11-26 08:27:06
飞碟探索(2016年5期)2016-05-10 23:44:30
饲料博览(2016年7期)2016-04-05 14:20:34
动物营养学报(2015年10期)2015-12-01 02:26:20
中国洗涤用品工业(2015年7期)2015-02-28 19:02:39
现代检验医学杂志(2015年4期)2015-02-06 02:02:11
