
松萝酸纳米混悬剂制备及其体内药动学研究
2022-07-22 08:44:52王奎鹏韩德恩
中成药 2022年3期
房 伟,王奎鹏,韩德恩
(1.河南中医药大学第一附属医院,河南 郑州 450000;2.河南中医药大学药学院,河南 郑州 450006)
松萝酸主要分布于松萝、石蕊衣、黄花岛等地衣植物中,资源丰富[1],具有抗病毒、抗炎、抗氧化、麻痹镇痛、抗肿瘤等活性[1-3],但该成分水溶性和脂溶性均较差[4],不利于药物溶解溶出及胃肠道透膜吸收,而且在体内容易受到各种酶的代谢作用[5],从而影响其口服生物利用度[4,6]。目前,对松萝酸已有脂微球[7]、自微乳[8]等制剂学研究,但处方组成及制备工艺较复杂;宋婷等[4]对该成分磷脂复合物及口服药动学进行了研究,但其黏性较大,不利于药物溶出,并且其口服生物利用度提高程度在109.67%~177.83%之间,具有较大的改善空间。
纳米混悬剂是通过稳定剂在某种制剂技术作用下,将难溶性药物制成胶态药物分散体系[9-10],可提高药物溶解度、溶出度、生物利用度,而且制备工艺简单。因此,本实验采用高压均质法制备松萝酸纳米混悬剂,并考察其体内药动学,以期为相关制剂学研究提供参考。
1 材料
EX125DZH型电子分析天平(美国奥豪斯公司);Mastersizer-300型粒度分析仪(英国马尔文仪器有限公司);安捷伦1200型高效液相色谱仪(OpenLAB CDS工作站,美国安捷伦公司);MYP13-2S型磁力搅拌器(上海梅颖浦仪器仪表制造有限公司);ATS型均质机(加拿大Seeker公司);LTB-800型超声仪(济宁鲁通超声电子设备有限公司);MDF-86V588D型超低温冰箱(中科都菱设备有限公司);H-600型透射电镜(日本电子公司)。
松萝酸对照品(批号190915,纯度98.9%,盛世康普化工技术研究院);松萝酸原料药(批号20191023,纯度97%,南京天诺新材料有限公司)。聚乙烯吡咯烷酮K30(PVP K30,批号191207,亚什兰集团公司);聚乙二醇1000维生素E琥珀酸酯(TPGS,批号20201005,西安海斯夫有限公司);大豆卵磷脂(批号190617,辅必成科技有限公司);吐温80(F1901016,上海雷允上药业有限公司);乳糖(批号D2101115,阿拉丁控股集团有限公司)。
2 方法与结果
2.1 HPLC法测定松萝酸含量
2.1.1 色谱条件 参考文献[5]报道,Welchrom C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇-[50 mmol/L KH2PO4-三乙胺(500∶1,超声混匀)](70∶30);体积流量1.0 mL/min;柱温35 ℃;检测波长285 nm。
2.1.2 线性关系考察 称取松萝酸对照品10 mg至量瓶中,加入5 mL三氯甲烷超声溶解,甲醇定容至50 mL,得200 μg/mL贮备液,甲醇制成10、5、1、0.5、0.1、0.02 μg/mL溶液,在“2.1.1”项色谱条件下进样测定。以松萝酸质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,得方程为Y=13.159 3X-0.813 5(r=0.999 8),在0.02~10 μg/mL范围内线性关系良好。
2.1.3 供试品溶液制备 取0.5 mL纳米混悬剂至100 mL量瓶中,加入80 mL甲醇超声溶解,室温静置30 min后定容,即得。
2.1.4 方法学考察 取供试品溶液适量,于0、3、6、12、18、24 h在“2.1.1”项色谱条件下进样测定,测得松萝酸峰面积RSD为0.46%,表明溶液稳定性良好。取同一份纳米混悬剂,按“2.1.3”项下方法平行制备6份供试品溶液,在“2.1.1”项色谱条件下进样测定,测得松萝酸峰面积RSD为1.70%,表明该方法重复性良好。取10、1、0.02 μg/mL对照品溶液,在“2.1.1”项色谱条件下各进样测定6次,测得松萝酸峰面积RSD分别为0.31%、0.25%、0.54%,表明仪器精密度良好。取0.25 mL供试品溶液至100 mL量瓶中,加入对照品溶液1.0 mL(低)、1.25 mL(中)和1.5 mL(高),在“2.1.1”项色谱条件下进样测定,测得平均加样回收率分别为99.07%、100.78%、100.26%,RSD分别为1.46%、1.02%、0.85%。
2.2 纳米混悬剂制备 采用高压均质法[10]。取松萝酸50 mg、大豆磷脂100 mg,加入10 mL丙酮,在50 ℃水浴下磁力搅拌2 h至溶液澄清,加到50 mL含有其他稳定剂的溶液中,减压旋蒸除去有机溶剂,补充蒸馏水至50 mL,在一定压力下循环均质数次,过0.45 μm微孔滤膜,即得。
2.3 粒径、PDI、Zeta电位的测定 取纳米混悬剂0.2 mL,加入20 mL蒸馏水稀释,混匀,取约3 mL至比色皿中,在粒度分析仪上测定粒径、PDI、Zeta电位。
李公甫家里有三口人,夫妻俩,外加一个小舅子。妻子是个妇道人家,只懂洗衣做饭操持家务;小舅子却满腹经纶,还酷爱钻研医书,但肩不能挑手不能提,目前还在家里吃闲饭。一家子都靠着李公甫来养活,他敢不尽心尽力地卖命工作吗?
2.4 单因素试验优化制备工艺
2.4.1 稳定剂种类 固定松萝酸用量50 mg,大豆磷脂用量100 mg,均质压力70 MPa,均质次数10次,考察PVP K30、泊洛沙姆188、吐温80、TPGS(用量均为100 mg)对粒径、PDI、Zeta电位的影响,结果见表1。由此可知,仅泊洛沙姆188所制备纳米混悬剂的Zeta电位绝对值超过30 mV,而且粒径、PDI最低,故选择其作为稳定剂。

表1 稳定剂种类对粒径、PDI、Zeta电位的影响(n=3)Tab.1 Effects of stabilizer type on particle size,PDI and Zeta potential (n=3)
2.4.2 稳定剂用量 固定松萝酸用量50 mg,大豆磷脂用量100 mg,均质压力70 MPa,均质次数10次,考察泊洛沙姆188用量60、80、100、120 mg对粒径、PDI、Zeta电位的影响,结果见表2。由此可知,不同用量所制备纳米混悬剂的PDI均小于0.3,表明纳米粒分布较为集中。当用量较小时,乳化能力可能不足而导致粒径稍大,但用量较大时粒径反而有所上升,可能是黏度增加所致;用量为100 mg时粒径较小,PDI最小,Zeta电位绝对值大于30 mV,故选择其作为稳定剂用量。
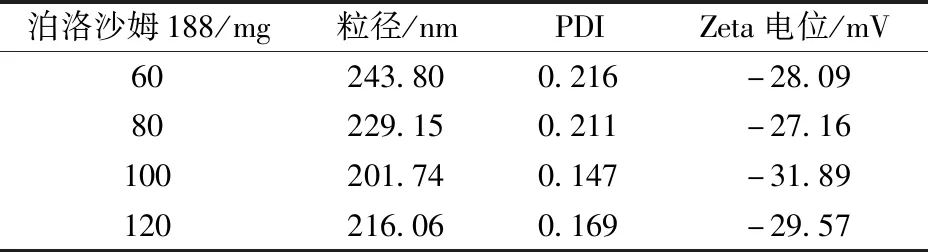
表2 稳定剂用量对粒径、PDI、Zeta电位的影响(n=3)Tab.2 Effects of stabilizer consumption on particle size,PDI and Zeta potential (n=3)
2.4.3 均质压力 固定松萝酸用量50 mg,大豆磷脂用量100 mg,稳定剂用量100 mg,均质次数10次,考察均质压力50、60、70、80、90 MPa对粒径、PDI、Zeta电位的影响,结果见表3。由此可知,随着均质压力增加,粒径有所下降,但在90 MPa时反而上升,并且PDI增加,可能是由于过大的均质压力会使纳米体系温度升高,导致纳米粒子之间发生聚集甚至融合,从而粒径增大,分布不均匀,故选择80 MPa作为均质压力。
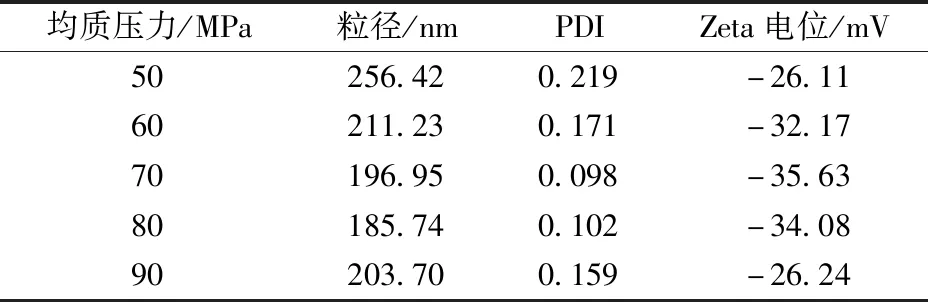
表3 均质压力对粒径、PDI、Zeta电位的影响(n=3)Tab.3 Effects of homogenization pressure on particle size,PDI and Zeta potential (n=3)
2.4.4 均质次数 固定松萝酸用量50 mg,大豆磷脂用量100 mg,稳定剂用量100 mg,均质压力80 MPa,考察均质次数6、8、10、12、15次对粒径、PDI、Zeta电位的影响,结果见表4。由此可知,随着均质次数增加,粒径逐渐下降,但在15次时粒径、PDI均有升高趋势,Zeta电位绝对值有下降趋势,故选择12次作为均质次数。
2.5 验证试验 单因素试验结果显示,最优制备工艺为稳定剂(泊洛沙姆188)用量100 mg,均质压力80 MPa,均质次数12次。按上述优化工艺平行制备3批纳米混悬剂,测定粒径、PDI、Zeta电位,结果见图1~2。由此可知,平均粒径为185.19 nm,PDI为0.089,Zeta电位为-34.08 mV,表明该工艺重复性较好。

图1 纳米混悬剂粒径分布Fig.1 Particle size distribution of nanosuspensions

图2 纳米混悬剂Zeta电位Fig.2 Zeta potential of nanosuspensions
2.6 形态观察 取适量纳米混悬剂,蒸馏水稀释50倍后滴至铜网上,1.5%磷钨酸钠负染,滤纸吸除多余液体,室温晾干后在透射电镜下进行观察。由图3A可知,纳米混悬剂呈球形或类球形,纳米粒之间无粘连。

图3 样品透射电镜图Fig.3 Transmission electron microscopic images for samples
2.7 冻干粉制备及溶解度测定 取纳米混悬剂适量,加入6%甘露醇并混匀,置于-50 ℃冰箱中预冻2 d后转到-20 ℃真空冻干机中2 d,即得冻干粉(图4),密封后于干燥器中保存。取适量,蒸馏水复溶后测得其平均粒径增加至231.42 nm,Zeta电位为-30.37 mV。

图4 纳米混悬剂冻干粉外观Fig.4 Appearance of lyophilized powder of nanosuspensions
再采用饱和溶剂法测定溶解度。取过量松萝酸、纳米混悬剂、物理混合物(比例同纳米混悬剂),置于蒸馏水中,平行3份,分别超声处理20 min至松萝酸不再溶解,置于25 ℃水浴中持续磁力搅拌2 d,取上层混悬液,6 500 r/min离心20 min,HPLC法测定上清液中松萝酸含量。结果,原料药在水中的溶解度为6.86 μg/mL,而纳米混悬剂为92.47 μg/mL,提高了13.48倍,另外物理混合物中其溶解度为12.62 μg/mL,可能是处方中表面活性剂的增溶作用所致,但提高程度远低于纳米混悬剂。
2.8 体外释药研究 取松萝酸、纳米混悬剂、物理混合物(比例同纳米混悬剂)适量(以松萝酸计均为15 mg),用超声脱气后的1%SDS溶液制成混悬液,并转移至活化透析袋中(截留分子量8 000~14 000 Da),以900 mL 1%SDS溶液为介质,设定溶出仪(配置自动取样器)温度为37 ℃,转速为100 r/min,于设定时间点各取样4 mL,并补充4 mL空白介质,6 500 r/min离心20 min,HPLC法测定上清液中松萝酸含量,绘制溶出曲线,见图5。由此可知,纳米混悬剂在60 min内累积溶出度达90.46%,90 min内基本溶出完全,但松萝酸在120 min内也仅为19.93%,另外物理混合物在一定程度上也增加了松萝酸溶出度,但程度远低于纳米混悬剂。
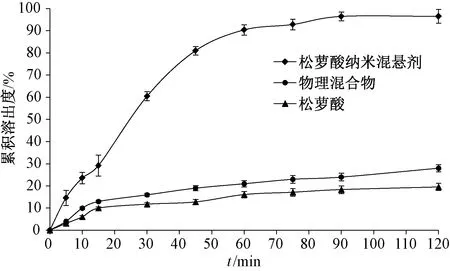
图5 松萝酸体外释药曲线(n=3)Fig.5 In vitro drug release curves for usnic acid (n=3)
2.9 药动学研究
2.9.1 分组、给药与采血 12只大鼠随机分为松萝酸组、纳米混悬剂组,每组6只,按15 mg/kg剂量灌胃给予相应药物的0.5%CMC-Na混悬液(以松萝酸计均为2.5 mg/mL),0.5 mL饮用水冲洗灌胃针后再次灌胃给药,于0、0.25、0.5、1、2、2.5、3、4、6、8、10、12 h眼眶后静脉丛采血各约0.2 mL,血样肝素化后立即3 500 r/min离心3 min,取上层血浆,置于-15 ℃冰箱中保存。另取6只大鼠,设置物理混合物组,同法灌胃给药。
2.9.2 血浆处理 参考文献[5]报道的方法,取含药血浆100 μL至离心管中,加入200 μL磷酸盐缓冲液(pH=7.4),密封后涡旋30 s,加入1 mL甲醇涡旋3 min,12 000 r/min离心10 min,分离上清液至另一离心管中,氮气缓慢吹干得残渣。加入100 μL甲醇后密封,涡旋30 s,进样测定。
2.9.3 方法学考察 取空白血浆适量,制成8.0、4.0、2.0、1.0、0.5、0.1 μg/mL血浆对照品溶液,各取0.5 mL,按“2.9.2”项下方法处理,在“2.1.1”项色谱条件下进样测定。以峰面积(Y)对松萝酸质量浓度(X)进行回归,得方程为Y=348.21X+50.11(r=0.992 9),在0.1~8.0 μg/mL范围内线性关系良好。
取空白血浆、含药血浆(纳米混悬剂给药12 h后)、0.1 μg/mL血浆对照品溶液适量,在“2.1.1”项色谱条件下进样测定,结果见图6,可知血浆内源性杂质未对测定产生干扰,表明该方法专属性良好。取8.0、2.0、0.1 μg/mL血浆对照品溶液,同一天内在“2.1.1”项色谱条件下进样测定6次,测得松萝酸峰面积RSD分别为3.15%、4.62%、4.19%,表明该方法日内精密度良好;同法于第1、2、3、4、5、6天各进样测定1次,测得松萝酸峰面积RSD分别为6.34%、8.17%、7.42%,表明该方法日间精密度良好。取含药血浆适量,室温下于0、2、4、6、12、24 h在“2.1.1”项色谱条件下进样测定,测得松萝酸峰面积RSD为7.53%,表明血浆在24 h内稳定性良好。取8.0、2.0、0.1 μg/mL血浆对照品溶液适量,在“2.1.1”项色谱条件下各进样测定3次,测得松萝酸平均加样回收率分别为90.42%、93.67%、87.07%。
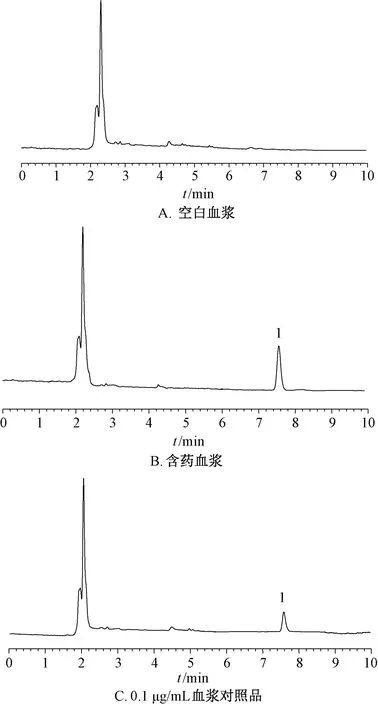
1.松萝酸1.usnic acid图6 松萝酸HPLC色谱图Fig.6 HPLC chromatograms of usnic acid
2.9.4 结果分析 绘制血药浓度-时间曲线,采用3P97程序统计矩模型计算主要药动学参数,结果见图7、表5。由此可知,与原料药、物理混合物比较,纳米混悬剂tmax缩短(P<0.01),t1/2延长(P<0.01),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相对生物利用度提高至3.09倍;物理混合物主要药动学参数与原料药比较有一定改变,可能与处方中稳定剂增溶作用有关,但差异均无统计学意义(P>0.05)。
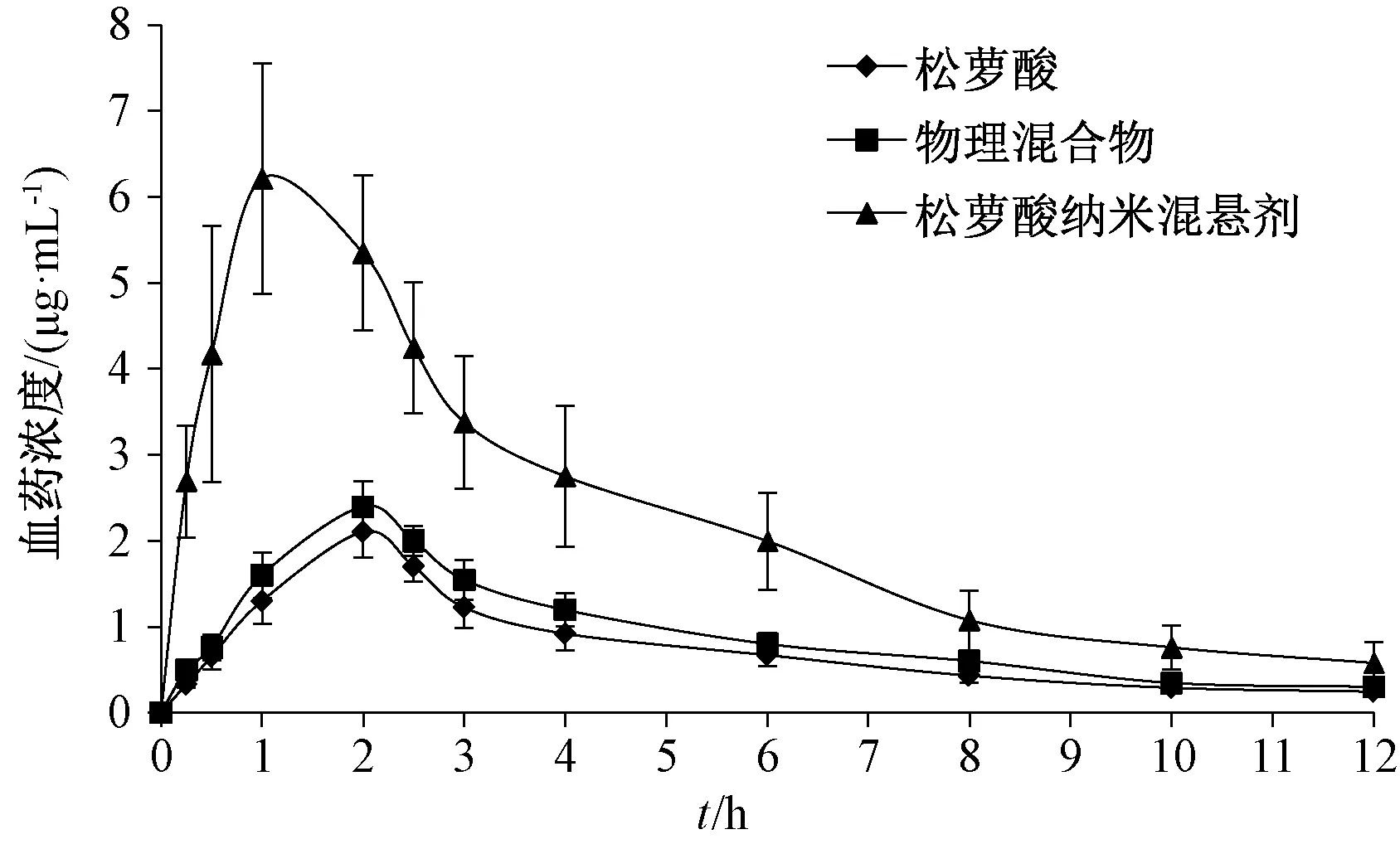
图7 松萝酸血药浓度-时间曲线(n=6)Fig.7 Plasma concentration-time curves for usnic acid (n=6)
3 讨论
本实验所用的泊洛沙姆188作为稳定剂,可提供较大的空间阻力,防止纳米混悬剂聚集[11-12]。另外,磷脂一方面发挥增溶作用,使药物形成均一溶液;另一方面可向纳米粒子提供电荷,从而增加了纳米粒子之间的排斥作用[13],提高了制剂的稳定性,与泊洛沙姆188共同发挥了稳定剂的作用。
表5 松萝酸主要药动学参数Tab.5 Main pharmacokinetic parameters for usnic acid
结果显示,松萝酸制成纳米混悬剂后溶解度、溶出度均大幅度提高,主要与该成分比表面积明显增加、稳定剂增溶作用等因素有关;纳米混悬剂tmax显著缩短,可能与该制剂促进药物溶出、提高药物前期溶出速率有关;文献[10,14]报道,纳米混悬剂可显著延长药物t1/2,可能是由于胃肠道环境较为复杂,影响因素较多,药物在胃肠道内完全溶出溶解需要一定时间,从而影响进入血液循环的时间所致,并且纳米药物能增加与胃肠道黏膜的粘附性,可能也会对上述药动学参数造成影响[14];口服生物利用度显著提高,这是因为将该成分纳米化后增加了其溶解度,促进了溶出。
研究表明,纳米混悬剂由于具有较小的粒径,可增加跨膜渗透性[9,15];稳定剂吸附于纳米药物表面,可降低胃肠道各种酶对药物稳定性的影响[9];纳米药物在伊派尔结中聚集[14-17],通过淋巴结中的M细胞进入体循环,从而改变吸收途径。另外,纳米混悬剂口服进入胃肠道后可能发生聚集,导致粒径变大,从而影响生物利用度提高程度。今后,本实验将对松萝酸纳米混悬剂的药效学开展研究[18],以期为该制剂开发提供更全面的资料。
猜你喜欢
昆明医科大学学报(2021年6期)2021-07-31 07:40:00
现代畜牧科技(2021年2期)2021-03-19 07:48:10
广西职业技术学院学报(2020年6期)2020-03-14 02:15:54
国外医药(抗生素分册)(2019年2期)2019-05-23 03:09:56
中成药(2017年4期)2017-05-17 06:09:50
数学物理学报(2016年5期)2016-08-24 07:38:38
油气地质与采收率(2014年6期)2014-12-16 17:45:18
保健与生活(2014年5期)2014-04-29 00:44:03
河南医学研究(2014年2期)2014-02-27 14:51:26
汽车与新动力(2012年4期)2012-03-25 10:09:38
