
膜电渗透装置分离戊二醛-水及电场对聚砜膜的影响
2022-07-22 09:59曹利娜纪志超
天津工业大学学报 2022年3期
王 韬,曹利娜,李 赛,纪志超
(天津工业大学化学工程与技术学院,天津 300387)
在膜分离的传统工艺中,以压力差作为传质推动力一直占有主要地位[1]。其主要原因是由于产生压力推动力的操作简便易行且动力来源简单。但如果从理论层面进行讨论,就会发现相比于压力,电场作为传质推动力更为强大。徐铜文[2]提出,在膜分离过程中,1/40 V 电场所提供的推动力相当于1 200 个标准大气压的压力推动力(假设浓度相为1)。
当前,以电场作为传质推动力的报道常见于汽车涂漆所用的膜-电泳工艺[3]、纳滤膜脱盐[4]以及电渗析去除金属离子[5-6]等工业。近10 年来,有关将膜电技术相结合用于膜生物反应器(MBR)过程,从而减轻膜污染、提高MBR 处理污水效率的报道也开始涌现[7-10]。传统的膜分离工艺中,渗透汽化(PV)是分离极性不同的有机/无机物液-液体系的常见技术。目前,PV 对物系分离促进作用的报道集中于对膜材料性能的改进[11-13]。其中,将电场应用于PV 过程的研究重点仍偏重于电效应对膜材料改性所产生的影响,而以电场本身作为PV 推动力的研究尚未有文献加以报道。
2015 年,以电势能差作为推动力用于分有离极性差异的物系开始见诸报道,在水-N2物系分离及强化膜蒸馏水中脱盐过程中,Du 等[14]和苗成朋等[15]采用平板-针状配置的电极所产生的电场(过程操作电压高达20 kV)对膜分离过程进行强化。而该研究中对应用最广泛且最常见的极性不同的液-液(有机/无机物)体系尚未涉及。该研究认为,分离极性不同物系的膜电装置不能采用成对的平板电极,否则无法提供足够的电场推动力。
本研究以相对弱化的电场(相对于参考文献[14-15])取代PV 中的温度场作为推动力,在自主设计的膜电渗透装置中应用成对平板电极进行实验,在近似匀强电场下建立较为完善的新工艺流程,使戊二醛-水物系经新型膜电装置处理后达到较好的分离效果,作为主要推动力的电势差相对于已知文献值成数量级降低,从而使新工艺能耗明显降低,实验的经济性与安全性显著提高,且整个膜分离过程中不产生相变。
1 实验部分
1.1 实验器材与设备
材料:聚砜(PSF)超滤膜,时代沃顿公司产品;超纯水,天津嘉华新宝水处理专业公司产品;戊二醛、苯甲醛,分析纯,上海腾准生物科技有限公司产品。
仪器:BR-400 型蠕动泵,淄博纽凯机电设备有限公司产品;2XZ-2 型旋片式真空泵,浙江台州求精真空泵有限公司产品;DW-P102-2ACEO 型高压直流电源,可变电压范围小于或等于1 000 V,东文高压电源股份有限公司产品;500 mL 规格的气体干燥塔、紫外分光光度计,上海翔雅仪器设备有限公司产品;膜电渗透装置,实验室自主设计。
1.2 膜电渗透装置的设计
本课题组在保持一定真空负压的条件下,附加近似匀强电场作为另一种推动力,对戊二醛-水物系进行无相变条件下的膜分离。由于钛基电极具有化学稳定性好、电导率高、电场发生效果优于普通金属电极等特点,因此将面积相等的网状钛基电极板置于膜组件两侧,并通过直流稳压电源产生近似匀强电场。按照上述思路所设计的膜电渗透装置的三维立体效果如图1 所示。
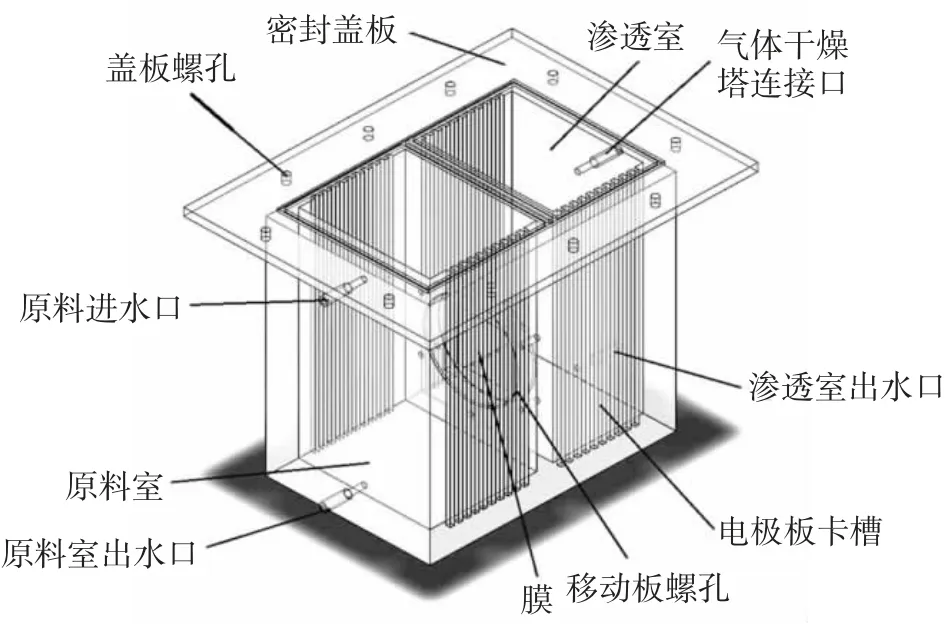
图1 膜电渗透装置的三维立体效果图Fig.1 Three-dimensional effect diagram of membrane electro-osmosis device
1.3 实验步骤
1.3.1 膜电渗透装置的密封安装
首先将两片电极板相对于膜片对称放置在两侧的原料室与渗透室,其中原料室为正极、渗透室为负极,调整电极板间距为10 cm。其次,将中央镂空的胶皮垫片置于两片有效膜面积一定的移动挡板之间,并用不导电的尼龙材质螺丝紧密固定胶皮垫片和移动挡板,每个螺丝孔都以热熔胶密封粘固。为了防止漏气,在盖板下面用胶皮垫片盖住,再用尼龙螺丝密封固定,最后用胶泥封住渗透室上方的导线连接孔以及渗透室和原料室两侧的出水孔。
1.3.2 实验装置的连接
实验流程如图2 所示。先将蠕动泵与原料室右侧上部的进水孔连接,然后将稳压直流电源正极接入膜电渗透装置的原料室,负极接入渗透室。为防止真空泵吸入水汽而对仪器内部造成损坏,因此需要在实验开始之前将渗透室左侧的上端口连接500 mL 的气体干燥塔,最后再将真空泵与气体干燥塔连接。

图2 实验流程图Fig.2 Experimental flow chart
1.3.3 实验原料注入实验装置
实验装置连接完以后,打开蠕动泵,进料速率调节保持在30~40 mL/min,然后将实验所需的原料液由原料液进水口注入到原料室当中。为了确保膜分离过程准确有效,在实验全程中原料液必须完全浸没膜。
1.3.4 真空抽吸
打开真空泵,实验过程中始终保持真空负压为-0.5 MPa,在压力达到稳定后计时30 min,计时结束后关闭真空泵,测量所得渗透液体积,并用紫外分光光度计分析渗透液组成。然后将渗透液再注回原料室。在0~1 000 V 电压且梯度为200 V 的条件下所得到的原料液渗透液依次用量筒收集测量体积并记录。为减小实验误差,同一电压条件下均3 次重复上述步骤,并将3次渗透液的测量体积取平均值记为V平均。
2 结果与讨论
2.1 纯水物系膜通量的测定
实验中膜的有效面积为16cm2,测量时间为30 min,根据膜通量的计算公式可得:

式中:J为膜通量(L/(m2·h));V为渗透液体积(L);S为有效膜面积(m2);T为测量时间(h)。
通过式(1)计算纯水物系的膜通量如表1 所示。

表1 纯水物系渗透液体积V 及膜通量J 的测定表Tab.1 Determination of permeable liquid product V and membrane flux J in pure water system
根据表1 绘制出电压与膜通量的关系图如图3 所示。

图3 纯水物系中电压对膜通量的影响Fig.3 Effect of voltage on membrane flux
由图3 可以看出,在纯水物系中,仅以真空负压作为推动力时,膜通量为2.3 L/(m2·h)。保持真空负压恒定,在施加电压后,随着电压的增大纯水体系膜通量增大,膜通量与电压成正相关关系,从而说明电压对纯水体系膜通量的提高有一定的促进作用。虽然水分子的极性强于大多数常见有机物,但由于其较大的介电常数,导致在本实验条件下水分子的极化作用减弱,表现为水分子受电场作用力随电压的升高而增加的较为缓慢,因而膜通量随着电压的增大增幅较小。
2.2 戊二醛-水物系膜通量的测定
通过关系式(1)计算,戊二醛-纯水物系的膜通量如表2 所示。

表2 戊二醛-纯水物系渗透液体积V 及膜通量J 的测定表Tab.2 Determination permeate liquid V and membrane flux J of glutaraldehyde-pure water system
根据表2 绘制电压与膜通量的关系如图4 所示。
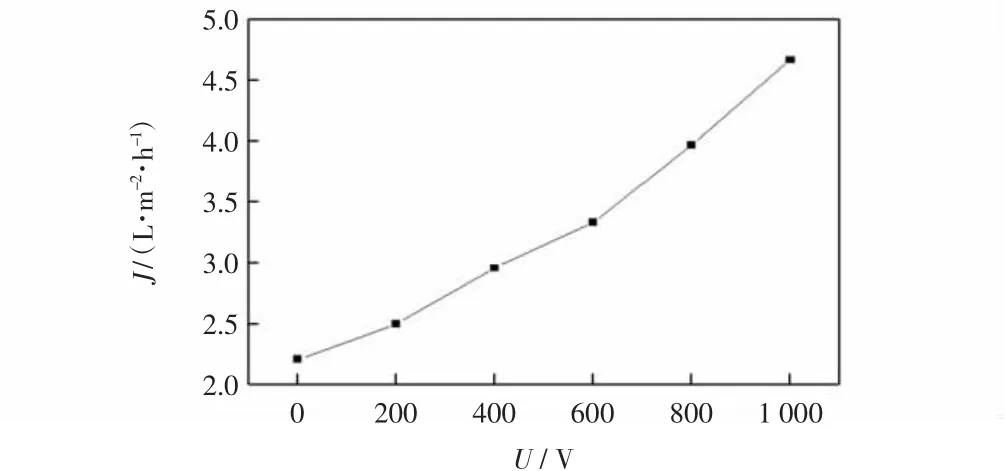
图4 戊二醛-纯水物系中电压与膜通量的关系图Fig.4 Effect of voltage on membrane flux in glutaraldehydepure water system
由图4 可以看出,在戊二醛-纯水物系中,在真空负压条件下,膜通量为2.2 L/(m2·h),按梯度施加电压后,与未施加电场时的膜通量相比,施加电场后的膜通量增大,因此电场的施加对戊二醛-纯水物系中膜通量有成正相关的影响。
2.3 戊二醛-纯水物系中戊二醛浓度的测定
通过用紫外分光光度计对所收集的戊二醛渗透液进行测试,所得戊二醛浓度如表3 所示。

表3 渗透液中戊二醛浓度Tab.3 Concentration of glutaraldehyde in permeate liquid
由表3 可知,戊二醛-纯水物系所得渗透液中戊二醛质量分数分布在8.27%~12.27%之间,原料液中戊二醛质量分数为5%,可知渗透液中戊二醛的含量扩大了1.6~2.5 倍,这是因为戊二醛相较于水分子更容易透过聚砜膜,但随电压不断增加,透过液中戊二醛浓度却不断降低,说明戊二醛分子较水分子透过聚砜膜的优势在逐渐降低。其原因是,随电场强度的增加,物质极性的不同对膜渗透效果产生的影响越来越明显。因为水分子极性明显强于戊二醛分子,故在电场作用下,水分子优先透过渗透膜,即极性差异在新工艺条件下成为主要影响因素。
2.4 电场对膜结构的影响
分别观察了戊二醛浸泡膜和戊二醛+电场(1 kV)膜的表面结构和断面结构,测试结果如图5、图6 所示。

图5 施加电场前后膜的表面形貌电镜图Fig.5 SEM of film surface before and after electric field

图6 施加电场前后膜的断面形貌电镜图Fig.6 SEM of membrane cross-section before and after electric field
图5(a)和图5(b)分别为上述2 样品的表面电镜图,图6(a)和图6(b)分别为上述2 样品的断面形貌电镜图。从图5 中可以清晰地看到,施加电场前后的两个样品的表面均是光滑、致密的,没有出现明显的分层的有孔结构,在施加1 000 V 的弱化电场作用下PSF 膜表面致密程度稍有提高,但形貌变化并不明显,这说明在该强度的电场条件下不会对聚砜膜的表面结构造成显著的影响。从图6 中可以看到,PSF 膜在施加电场前后的截面结构上发生了一些变化,其变化主要表现在戊二醛+电场实验中的PSF 膜截面出现了一些横向孔结构,该横向孔的出现直接影响了聚砜膜的亲水性的大小。本实验结果表明,在电场作用下,原本处于连通状态的指状膜孔由于横向孔结构的影响而发生中断,进而使得PSF 膜的亲水性降低。Darestani等[16]曾在2 000 V 电压和90 ℃的实验条件下对PVDF微滤膜进行测试,其SEM 结果表明,因为施加了高电压电场,PVDF 的内部结构由膜横截面上的膜孔结构方向一致、膜孔间隙紧密、膜内孔分布相对均匀且相互连通,转变为膜孔隙分布不均匀、层次分明的状态。Darestani 等认为出现这种改变的原因是由于高电压电场的作用,原本排列无序的含氟基团沿电场方向相对有序地排列旋转。这种相对有序的排列旋转使膜内部的α 晶形主体结构消失,并且全部转变成为β 晶形主体结构。武利顺等[17]也研究发现PVDF 的α 晶型在室温下条件下结构是稳定的,但是机械变形以及高压电场作用下可将α 晶型转变为β 晶型,并且只有β 晶型驻极体才显示出强的压电性能。
在本课题组实验所得的膜SEM 图中却发现PSF膜的内部膜孔结构的变化不像Darestani 等所描述的那样明显,除了考虑本实验所用电压远低于文献[16]中所达到的2 kV 以外,更主要的原因是本实验的整个实验过程都是在室温条件下进行的,较低的温度难以改变膜内部的链节强度,因此对膜结构的影响较小。
2.5 电场对膜亲水性的影响
表4为不同情况下PSF 接触角。

表4 不同情况下PSF 的接触角Tab.4 Contact angle of PSF under different conditions
由表4 可知,上述PSF 膜的接触角均在55°~70°之间,这表明膜的亲水性能较好。实验数据的结果表明,纯水物系和AR 级戊二醛物系,在施加1 000 V 电压的电场后,聚砜膜的接触角与未施加电场的情况相比增加8°~10°,因此施加电场可以使得聚砜膜的亲水性降低,该结论也与前文中SEM 测试的结果一致。
2.6 电场对官能团的影响
图7所示为不同的情形下PSF 膜的FTIR 图。

图7 不同条件下PSF 膜的红外光谱图Fig.7 Infrared spectra of PSF membranes under different conditions
由图7 可知,各种情况下均在1 140 cm-1处为砜基(—SO2—)中的S—O 键的对称伸缩振动吸收峰,1 330 cm-1处为该砜基(—SO2—)中的S—O 键的不对称伸缩振动吸收峰,1 500 cm-1和1 590 cm-1处的峰是聚砜的链节结构中苯环骨架的特征吸收峰,1 720 cm-1处为醛基中C—O 键的伸缩振动吸收峰。图7 中,在戊二醛+电场实验中1 720 cm-1处多出一个醛基中C—O 键的强伸缩振动吸收峰。该吸收峰的出现通常是由于强吸电子基团的存在,羰基中的C 原子的电性增强,进而使C—O 双键键间吸引力增大以及键力常数K增大所致[18-20]。但是在本实验中,溶液中仅存在戊二醛,而不存在其它强吸电子基团,这说明弱化匀强电场的施加产生了与强吸电子基团存在时同样的效果,从而使醛基中C—O 键的伸缩振动出现了吸收峰,而对类似现象的相关报道在以往文献中尚未发现。由于C—O键属于极性共价键,而该键的存在不会改变物体的平动状态[21-22],如文献所述,虽然正负电荷受电场力的作用而发生转动,但极性分子在电场存在条件下产生的力偶距在其作用面的任意点合力为零,实验结果表明,一定强度电场的存在能够导致某些物质极性增强。为了验证电场对醛类物质官能团结构变化的影响,用苯甲醛+电场进行对比实验,由图7 可见,在1 720 cm-1处同样出现了C—O 键的伸缩振动吸收峰,故上述结论得到验证。该FTIR 图说明了膜的分子结构不会因为弱化匀强电场的存在而发生明显的变化,但能够通过引发物系中的醛类化学官能团结构变化从而强化膜渗透过程中的极化作用,成为新工艺过程中的主要推动力。
3 结 论
本实验通过使用本课题组自主设计的膜电渗透装置,仅在0~1 kV 弱化匀强电场条件下,分离戊二醛-纯水物系,并且通过膜通量、接触角、SEM、FTIR 等测试方法进行表征,研究结果表明:
(1)施加电压引入电场后,在亲水性降低的情况下,纯水和戊二醛-纯水物系的膜通量仍有所增加。
(2)施加电压后戊二醛-纯水物系的膜通量相较于单一纯水物系的膜通量有所减少。由于水的极性远大于戊二醛的极性,从而证明在膜电渗透过程中电场下的极化作用是膜分离的主要推动力,其强化效果甚至能够使由于电场造成的膜亲水性的降低可以几乎完全忽略不计;且电场对物系中极性分子的作用力随电压的升高而不断增大。
(3)渗透液中戊二醛质量分数分布在8.27%~12.27%,相比于原料液中5%的戊二醛初始含量,渗透液中戊二醛的含量扩大了约1.6~2.5 倍。
(4)在0~1 kV 的弱化电场下,电场的施加使膜的亲水性降低,接触角减小。
(5)一定强度的弱化匀强电场的施加能够产生出与强吸电子基团存在时同样的效果,表现为使醛基中C—O 键的伸缩振动出现了吸收峰,即电场作用下的醛类物质极性得到增强。
猜你喜欢
中学生报·教育教学研究(2022年1期)2022-04-18
汽车实用技术(2020年5期)2020-04-10
现代盐化工(2019年4期)2019-09-10
时代英语·高一(2019年5期)2019-09-03
青年时代(2017年2期)2017-02-16
新教育时代·教师版(2016年31期)2016-12-07
新教育时代·教师版(2016年30期)2016-12-05
时代青年(上半月)(2009年2期)2009-08-11
数理化学习·高一二版(2009年2期)2009-03-30
数理化学习·高一二版(2009年1期)2009-03-19
