
Ⅳ期结直肠癌患者淋巴结转移相关基因的研究
2022-07-19 02:11:58骆泽民方建惠韦良宏陈海东易廷庄
右江民族医学院学报 2022年3期
骆泽民,方建惠,韦良宏,陈海东,易廷庄
(1. 广西钦州市第一人民医院消化内科,广西 钦州 535000;2. 广西钦州市第一人民医院肿瘤内科,广西 钦州 535000;3. 右江民族医学院附属医院,广西 百色 533000)
结直肠癌(colorectal cancer,CRC)是消化到最常见的恶性肿瘤之一,根据最新的流行病学调查的研究发现,每年在全球范围内的新发病例超过了100万例,更重要的是超过一半的患者确诊时已经到了晚期[1]。淋巴结转移是晚期CRC最常见的转移方式[2],同样也是CRC患者预后的决定性因素之一[3]。具体说来,对于没有淋巴结转移的患者,术后的5年生存率甚至超过了80%,而有淋巴结转移的患者即便没有远处转移的征象,术后的5年生存率下降到了约55%[3-4]。然而,目前对晚期CRC的淋巴结转移的关键机制尚不明确,这制约了进一步对CRC淋巴结转移针对性的临床进展。为此,本研究通过生物信息学方法对可切除的远处转移性结直肠癌(Ⅳ期)淋巴结状态进行分析,并对原发病灶的基因表达谱进行运算,通过一系列的方法探索影响Ⅳ期CRC患者淋巴结转移相关的核心基因,为进一步对转移相关的潜在机制的探索提供线索。
1 方法
1.1 表达谱芯片下载及分析 本研究通过对GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)进行检索,检索关键词包括“colorectal cancer”,“CRC”,“lymph node”,“metastasis”等,最终选择GSE63596的表达谱芯片进行后续研究。GSE63596数据集采用了GPL17077平台文件,纳入了15例可切除的Ⅳ期CRC患者,其中7例术后病检提示淋巴结阴性,8例为淋巴结阳性。男性10例、女性5例;年龄范围在45~80岁之间。采用Agilent微阵列技术对所有15例患者的mRNA谱进行分析,差异分析采用LIMMA包进行运算,统计学过滤条件为:|Log2FC|>2,P<0.05。
1.2 淋巴结转移风险模型 本研究通过对术后淋巴结的状态以及原发病灶的表达谱数据进行分析,采用加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)方法筛选出与CRC淋巴结转移相关的靶基因并构建预测模型。首先分别计算了每个基因的标准差(standard deviation,SD),剔除了SD最小的前25%的基因,去除了离群的基因和样本,随后通过主要连接关系和Pearson相关矩阵建立两个基因之间的相关矩阵。并通过对网络拓扑结构的分析,确定软阈值大小和相应的平均连通性。 拓扑重叠矩阵(topological overlap matrix,TOM)用于度量一个基因的网络连通性,为了将表达谱相似的基因分类到基因模块中,根据基于TOM的差异测度进行平均连锁层次聚类。为了进一步分析模块,计算模块特征基因(MEs)的差异性,通过Pearson检验来评估MEs与淋巴结转移的相关性,以确定相关模块。选择与转移高度相关的模块作为淋巴结转移模块进一步进行分析。
1.3 蛋白互作网络构建及LASSO回归分析 选择与淋巴结转移风险最高模块进行分析。利用STRING数据库(https://string-db.org/)进行蛋白-蛋白的互作(PPI)分析,删除没有互作关系的蛋白。同时利用R软件的LASSO回归分析包筛选转移模块中基因。logrank用于检验KM生存分析比较两组之间的生存差异,进行了timeROC分析以比较预测模型的准确性和风险评分。LASSO回归算法进行特征选择,采用10倍交叉验证,以上分析采用R软件包glmnet。对于Kaplan-Meier曲线,P值和95%置信区间(CI)的危险比(HR)通过logrank检验和单变量Cox比例危险回归得出。
2 结果
2.1 差异基因分析 差异基因分析采用R软件LIMMA包进行筛选,基于|Log2FC|>2,P<0.05。为阈值筛选GSE63596数据集中淋巴结阳性与淋巴结阴性的原发性肿瘤的差异基因。根据相应平台注释信息,统计将探针ID转换为gene symbol,如涉及多个探针对于同一个gene symbol时,取多个探针对应的平均值。分析之前采用normalize.quantiles函数进行数据标准化处理,以及主成因分析(principle component analysis,PCA)的预处理。针对数据预处理结果,通过箱线图进行评估数据标准化情况(见图1A);数据批次效应情况通过对比批次去除前后可视化PCA图进行评估(见图1B、图1C)。差异分析的结果提示有501个基因在淋巴结阳性的肿瘤中表达上调,另外有102个基因表达下调。差异表达基因(differentially expressed genes,DEGs)的具体分布采用火山图(见图1D)以及聚类分析的热图(见图1E)进行可视化展现。
为探索淋巴结转移的相关分子机制和潜在的信号通路,将DEGs进行GO功能富集以及KEGG信号通路富集分析。结果提示在淋巴结阳性的肿瘤组织中cAMP信号通路被显著激活,而对白介素1的反应受到了显著抑制(见图2)。然而DEGs的富集分析虽然在一定程度上提示了淋巴结转移的相关机制,但是这其中存在很多不相关因素的干扰。因此,还需要进一步分析并筛选出淋巴结转移的核心功能分子。
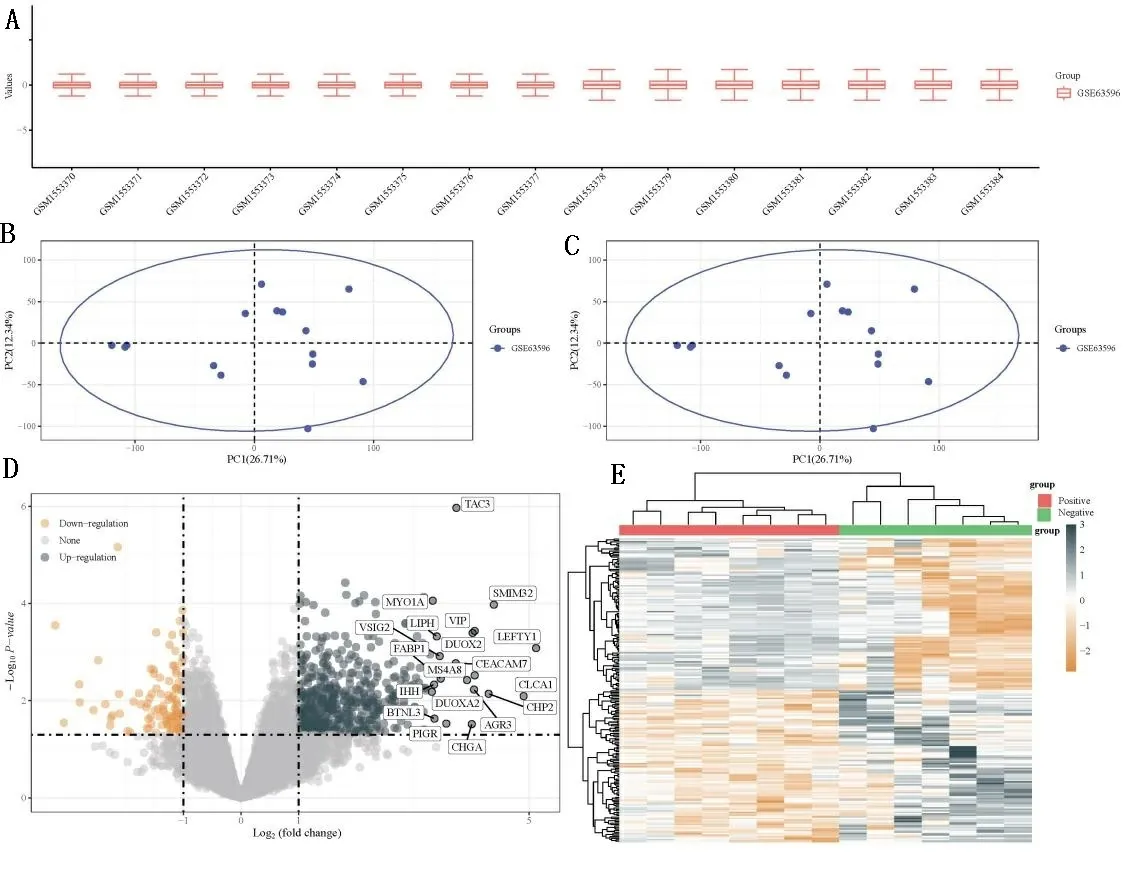
A: 数据标准化后箱线图。B、C: PCA结果图;D:差异基因火山图;E:差异基因表达热图
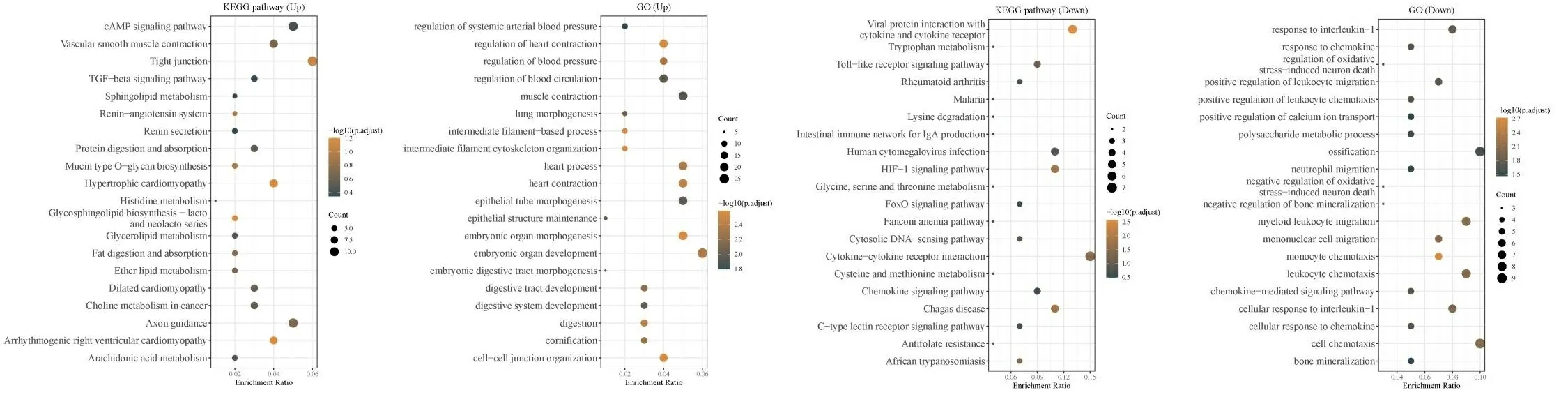
图2 差异表达基因富集分析
2.2 基因模块与临床性状关联分析 本研究通过构建WGCNA模型对经过处理后的数据进行分析,确定了软阈值(β);β 值的一般范围在1~20之间时,本研究中 β=20 (见图3A)以及相应的平均连接度= 0.12 (见图3B)。
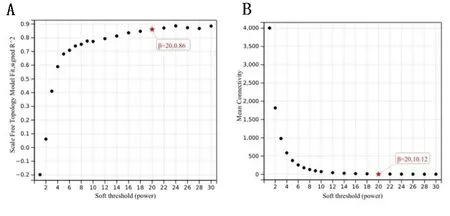
图3 WGCNA模型的软阈值(β)以及平均连接度
通过R语言软件中cutreeDynamic和moduleEigengenes函数绘制聚类图,去除相似模块的影响,共得到15个模块(见图4A),每种模块间的相关性(见图4B)。需要分析的主要变量参数为淋巴结转移的状态,方块中分别显示了每个模块与其的相关性系数及其P值(见图4C),结果发现淋巴结阳性与drakgrey模块显著相关(cor=0.64,P<0.01)。为了进一步分析模块,计算模块特征基因(MEs)的差异性,通过Pearson检验来评估MEs与淋巴结转移的相关性,以确定相关模块,结果也证实了drakgrey模块的基因与淋巴结转移的强相关性(r=0.53,P<0.01),见表1、图5A。将drakgrey模块中的DEGs提取出来,构建PPI网络后刷选出8个Hub基因(见图5B)。
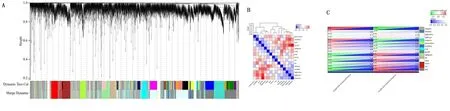
图4 WGCNA模型的核心模块筛选
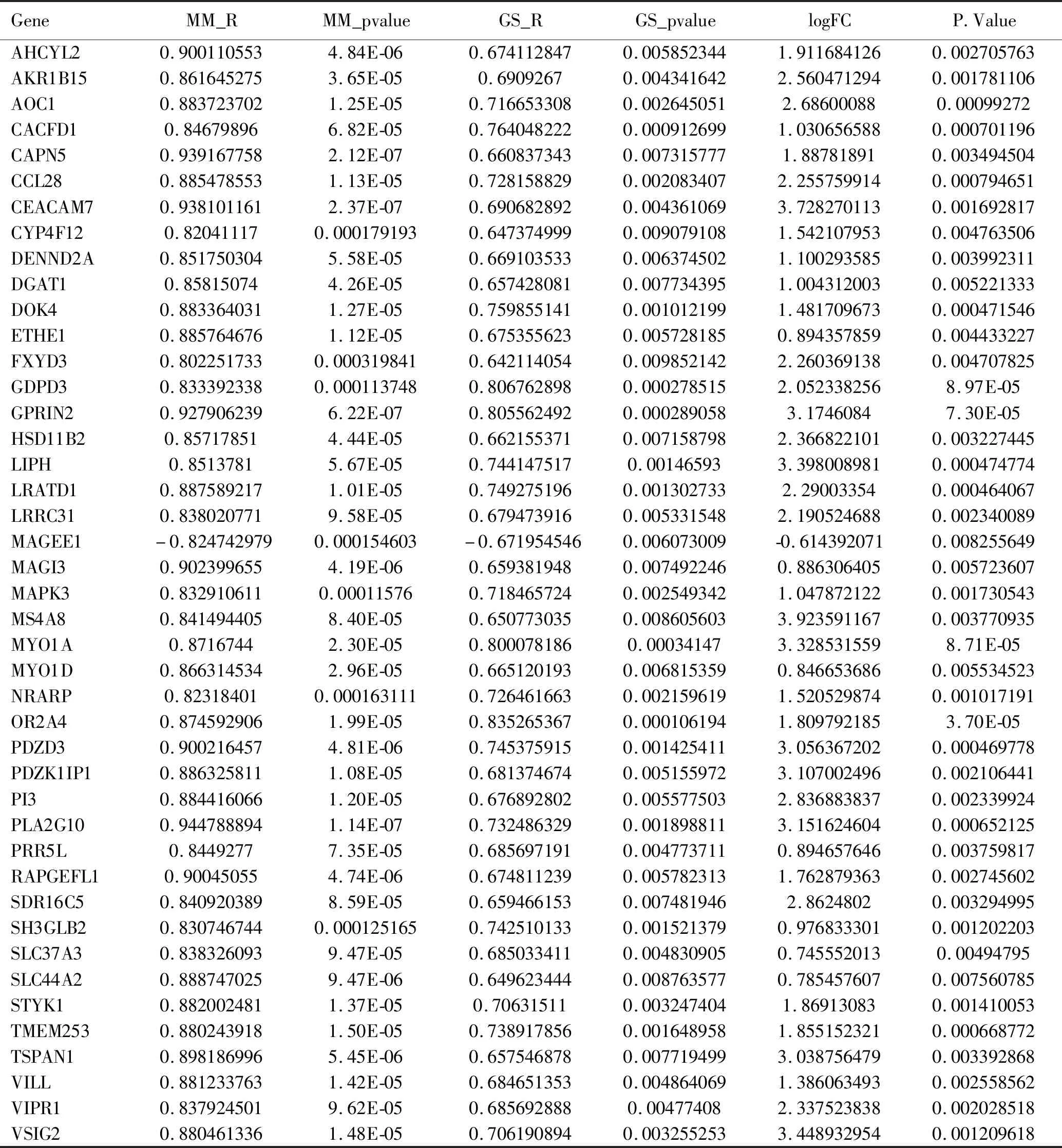
表1 drakgrey模块与淋巴结转移的相关的基因
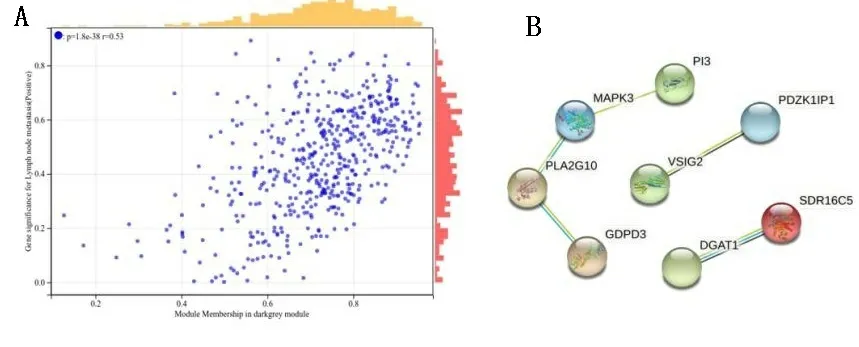
图5 淋巴结转移的核心模块与核心基因
2.3 LASSO 回归分析 为确定上述方法筛选出的Hub基因对预后的影响,基于TCGA数据库,下载Ⅳ期结直肠癌的表达谱数据和临床相关数据作为验证级。在本研究中,使用R软件包glmnet,整合生存时间、生存状态和基因表达数据,利用LASSO回归分析,以获得最优模型。本研究设置Lambda值为0.0407,获得基因4个(DGAT1、GDPD3、PDZK1IP1、PLA2G10),见图6,构建的模型公式为Riskscore=(0.1454)*DGAT1+(-0.0684)*GDPD3+(0.1404)*PDZK1IP1+(-0.1724)*PLA2G10。
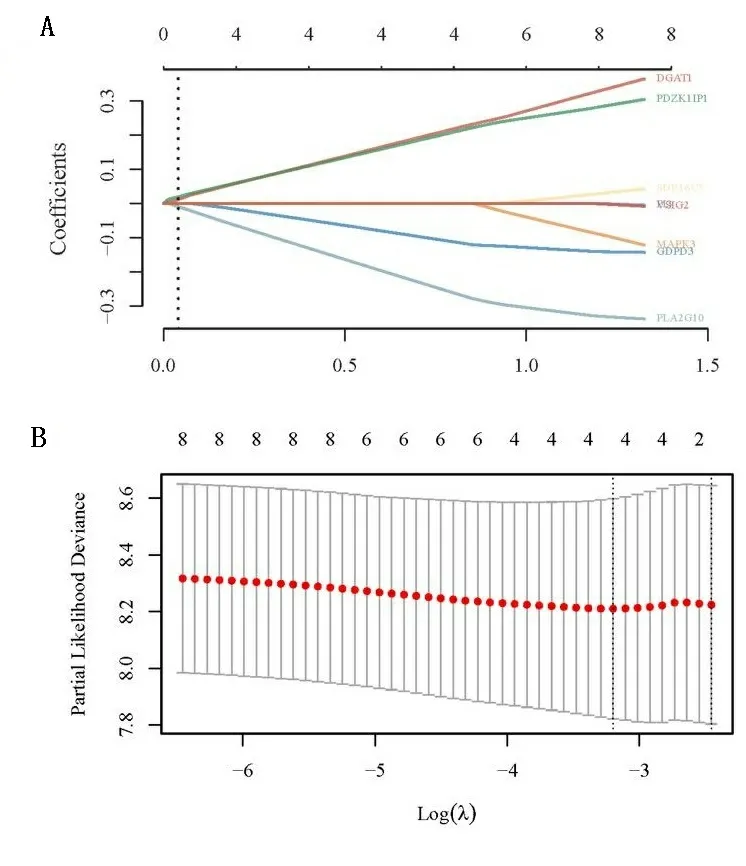
图6 LASSO 回归分析筛选Lambda值以及构建Riskscore模型
基于LASSO 回归分析的构建Riskscore模型评分,分为高风险组和低风险组,进一步使用“pheatmap”程序包绘制风险曲线图和生存状态图。可见参数基因在高危风险组的预后明显短于低风险组(HR=2.63,95%CI:1.857~3.1112,P=0.036)。ROC分析用于模型的构建过程中对结果预测的性能的一个评估。本研究中的预后模型分别绘制了1、3、5年OS的ROC曲线,结果提示该模型具有较强的预后判断价值(见图7)。
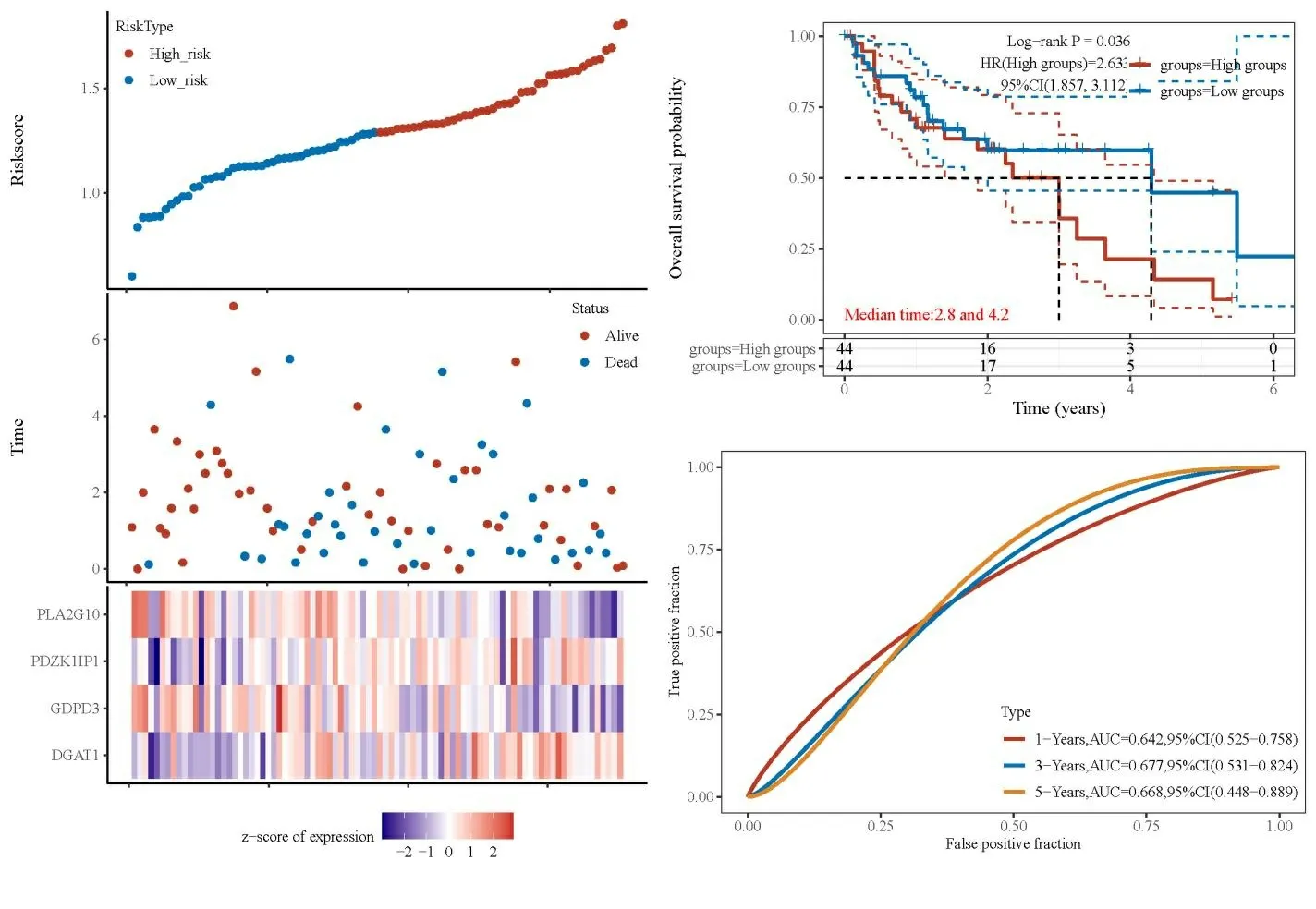
图7 Riskscore模型的预后预测价值
3 讨论
CRC的淋巴转移的发生机制目前仍旧存在争议,随着高通量技术的发展以及生物信息方法的应用,使得人们对病因以及机制的研究有了新的方法。本研究通过WGCNA以及LASSO 回归分析最终确定了4个与淋巴转移最为相关的Hub基因(DGAT1、GDPD3、PDZK1IP1、PLA2G10),并基于其表达水平构建了Riskscore评分的预测模型。
DGAT1是一种表达于内质网的多膜蛋白。它可以通过将酰基辅酶A转化为甘油三酯来合成甘油三酯,甘油三酯在脂质合成中起着关键作用[5-6]。DGAT1是MBOAT家族的成员,该家族的其他成员同样在脂质代谢、信号转导和激素治疗中发挥重要作用[7]。有研究认为DGAT1参与肿瘤细胞中IL-8表达的负调控[8]。IL-8可通过Wnt/β-catenin途径介导的EMT促进肿瘤的迁移[9]。 GDPD3在维持CML干细胞静止和TKI抵抗中起重要作用[10-11]。另外,在肌层浸润性膀胱尿路上皮癌患者中,GDPD3的表达水平还能与新辅助化疗的反应具有相关性[12]。但是尚未有足够证据表明,GDPD3可以直接参与了肿瘤的转移过程。PDZK1IP1是一种分子量比较小的蛋白(~17 kDa),在多种癌症中上调,虽然PDZK1IP1没有酶活性或转录活性,但它通过调节多种细胞信号通路发挥其所描述的作用。PDZK1IP1过表达激活Notch通路,从而使该通路更为活跃[13-14]。此外,PDZK1IP1的过表达可以通过增加活性氧(ROS)减少NFκB活化和细胞自噬[15]。ROS活性的增加可以提高基因突变率和基因组的不稳定性[16-17]。有研究报道[18],PLA2G10的过度表达促进了SK-LMS-1细胞的生长和G1/S转换,反之,PLA2G10的敲除减缓了细胞生长并导致G1期阻滞。而这种现象在CRC中同样被观察到,且被认为与炎症的发生密切相关[19-20]。
在本研究中,筛选取4个基因构建了转移相关的预后模型,并绘制风险曲线及热图,希望从基因角度对预测CRC患者的预后,以期为CRC的发生、发展及转归的生物学过程研究提供依据。但不可否认的是,本研究结果还需进一步在临床样本中获得更为可靠的验证,以及通过体内/外实验验证上述基因的临床和生物学意义。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
成都信息工程大学学报(2022年3期)2022-07-21 09:35:30
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01 06:27:42
天津医科大学学报(2021年4期)2021-08-21 02:14:40
心电与循环(2020年1期)2020-02-27 07:48:24
养生保健指南(2019年11期)2019-12-17 08:18:52
文理导航·教育研究与实践(2015年12期)2015-12-04 00:49:23
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:36
湖北农业科学(2014年3期)2014-07-21 10:25:00
中国中西医结合外科杂志(2013年3期)2013-03-11 20:05:01
