
一次性使用负压引流护创材料包的无菌保证与细菌内毒素质量风险评估
2022-07-16 05:11李发家徐红梅
大众科技 2022年5期
李发家 徐红梅 王 亮
一次性使用负压引流护创材料包的无菌保证与细菌内毒素质量风险评估
李发家1徐红梅1王亮2
(1.苏州凯瑞斯生物科技有限公司,江苏 张家港 215600;2.江苏艾迪尔医疗科技股份有限公司,江苏 张家港 215600)
文章对一次性使用负压引流护创材料包(VSD)的无菌保证和细菌内毒素的质量风险进行评估分析,通过分析造成灭菌前产品初始污染菌/生物负载污染的风险因素,以及对风险因素的评价并采取一定降低风险的控制措施,来评估产品的实际风险控制水平。通过分析数据,制定合理的控制依据,使得风险控制在可接受范围内,证明产品的无菌保证和细菌内毒素质量风险是完全可以控制的。
一次性使用负压引流护创材料包;无菌保证;细菌内毒素;初始污染菌;生物负载
引言
无菌医疗器械是指一种没有存活的微生物的产品。在生产无菌医疗器械时,其灭菌前产品的生物负载以及细菌内毒素检测是无菌产品质量控制程序中密不可分的部分。医疗器械生产企业需按照各自生产的产品特性对医疗器械产品上的生物负载数据进行采集,制定生物负载监测计划并定期进行评估,同时运用统计学方法原理,制定相应的微生物污染控制水平,通过该方法的应用,将医疗器械生产过程中的微生物污染风险降到尽可能低的水平,使产品灭菌后细菌内毒素控制在一定范围之内,以验证其生产流程能持续满足既定的控制要求,确保产品符合预期设定要求,指导生产实践[1]。
一般来说,通过比较和评估设定的限值(警戒限和纠偏限)和产品上的微生物检测值,来判断设定的限值是否合理。警戒限和纠偏限体现了生产作业过程中的风险控制要求。限值是根据在生产过程中正常运行操作条件下收集的数据而确定的。警戒限和纠偏限有助于监控生产过程是否连续。通常情况下,超出警戒限表示产品质量(微生物风险)可能已偏离基线,如果超过了纠偏限,则意味着清洁生产条件已失控,产品受微生物污染的风险较高。文章所述质量风险评估方法适用于对VSD产品的无菌保证与细菌内毒素控制水平的质量风险评估与控制方法[2]。
1 术语和定义
菌落形成单位CFU:微生物在经过培养之后,由单个菌体或聚集成团的多个菌体在固体培养基上生长繁殖所形成的集落,称为CFU[3]。
修正系数:用于补偿不能从产品和/或微生物培养中完全采集到的数据[3]。
生物负载:产品和/或无菌屏障系统表面或内部存活微生物的总数[4]。
合格限(即行动线):该标准可以是国家药典、法规标准,或者是通过验证得出数据来证明系统处于稳定状态时的限值之内的区域范围。合格线又被称为行动线,即当达到这个限度时,为了防止系统出现不合格时造成损失,需要对系统进行处置,如果超过限额,则被视为不合格。
警戒限:通常由趋势分析确定,基于系统性能水平和测试数据,其限度可以是企业内部控制质量标准。如果数值超过此限制,则表明系统可能存在问题,但不需要进一步处理,可能需要额外增加相对应的监控项目或检测频率。设置警戒限旨在提示系统注意恶化的可能性,并应密切监测以便及时采取行动[5]。
纠偏限:指的是限值在警戒限和行动限之间的区域,系统处于高风险状态,可能随时出现不合格,所以有必要采取一定的预防纠正措施[6]。
2 风险评估
2.1 风险因素分析
无菌产品的质量保证侧重于产品的无菌保证和细菌内毒素的控制。在无菌医疗器械的整个制造过程中,因医疗器械种类的多样性和复杂性,在标准制造条件下生产的产品仍可能受到微生物的污染。本项目的最大潜在风险因素在于VSD的组成部件较多、整体体积较大,其中创面敷料是直接接触创面伤口的,敷料体积比较大,从厂家出厂之后,到本厂来料检验时,以及再到最终产品灭菌前的初始污染菌数量,直接影响最终的灭菌效果以及内毒素的风险。若创面敷料中含有大量微生物,会造成灭菌前微生物无法控制进而超出灭菌工艺的能力,威胁产品的无菌保证水平。
此外,在成品检验过程中创面敷料的细菌内毒素主要来自于原材料敷料中已有的内毒素和敷料中微生物繁殖的代谢产物。换句话说,就是只要控制灭菌前产品上的微生物总量,就能控制相对应的细菌内毒素风险。
2.1.1影响产品生物负载的因素
VSD灭菌前生物负载的影响因素种类较多,根据YY/T0316标准[7]识别影响其生物负载的风险源,包括但不仅限于以下五点:
(1)原材料及包材中的微生物;
(2)环境条件;
(3)设备和工作台面;
(4)人员与生产操作;
(5)灭菌前产品生物负载的控制标准。
2.1.2影响创面敷料细菌内毒素的因素
影响创面敷料细菌内毒素的因素有:
(1)原材料以及包材自身携带的细菌内毒素;
(2)创面敷料中能产生细菌内毒素的活微生物,在灭菌前繁殖代谢产生。
2.2 风险因素分析评价与控制策略
2.2.1微生物的污染评价与控制策略
通常情况下,产品在灭菌前都存在一定程度的微生物污染,其污染程度用校正后平均生物负载cfu/g、cfu/SIP或cfu/cm2来表示。产品微生物污染的影响因素主要有如下方面。
2.2.1.1原材料和包装材料中的微生物
首先,对原材料和包装材料的生产企业在生产、质量控制、仓储、运输、验收等方面的质量可控性进行了详细的评估。因为存在于原材料和包材中的微生物可能会有进入产品的风险[8]。公司对原材料以及初包装材料均制定了内控微生物限度标准,以减少灭菌前微生物的总量,从而降低最终生物内毒素的风险。

表1 VSD的微生物限度标准
在没有历史数据的情况下,按照YY/T1737-2020医疗器械生物负载控制水平的分析方法附录B的方法建立纠偏限和警戒限,以创面敷料为例,技术要求中最大规格为300 mm×200 mm×30 mm,约为60 g,纠偏限设置为VDmax25允许的最大值1000 cfu/SIP,警戒线设置为60%的纠偏限,即为600 cfu/SIP,因创面敷料的规格比较多,大小体积不一样,重量也不一样,故评定标准限度定为以每克微生物的限度比较合理,即为10 cfu/g。同理,其他原材料以及初包装材料所定的微生物限度要求远远严格于警戒线,在原材料以及包材来料检验时,初始污染菌的检测值符合所定标准,也远远低于微生物限度值。
需要注意的是,在正常生产之后,基于一定数量的原材料来料检验生物负载数据分别进行警戒限和纠偏限的重新评审并形成文件,以确保其持续适用性。
2.2.1.2环境条件
VSD的生产过程包括两个过程:一是原材料的厂家生产过程;二是按照生产厂家的工艺流程进行生产的过程。在生产过程中存在有可能来自于生产环境中的微生物污染的风险,为了控制上述风险,创面敷料、医用贴膜、引流管、延长管、冲洗管均是厂家在十万级洁净环境条件下生产的。加强洁净环境的有效控制有利于进一步降低产品污染的风险。
在本公司洁净车间验收初期,由具有资质的第三方环境检测机构对洁净环境以及空调系统进行全性能检测,检测结果均符合《无菌医疗器具生产管理规范》YY0033-2000[9]以及《医药工业洁净厂房设计规范》GB50457-2019[10]中的规定,之后检验人员也做了对洁净车间全性能连续监测、洁净车间不连续生产验证、消毒剂消毒效果验证、洁净工作服清洗验证、工艺用气验证等均符合要求。工作环境和污染控制程序中规定了洁净车间空调系统的日常监测项目以及监测频次。从空调净化系统和洁净环境验证及日常监控数据来看,空调系统和洁净环境均能符合设计要求,为无菌产品的生产提供稳定的十万级生产环境。在正常情况下每年邀请第三方具有资质的专业机构对洁净环境进行全性能检测,并出具合格报告。
2.2.1.3设备和工作台面
在VSD原材料半成品进入洁净环境之后,部分用到清洗、烘干、包装封口等设备。设备以及工作台面存在残留微生物的可能,对在生产过程中的产品的生物负载存在潜在风险。认真执行清洁消毒作业,日常清洁采用新洁尔灭/恩杜罗消毒液擦拭设备和工作台面,按照洁净区手和物体表面细菌计数检测规程进行日常监测,检测结果应符合要求。
2.2.1.4人员与生产操作
在洁净环境中最大的污染源来自于人员及其活动。一方面,人员对洁净环境的污染间接影响了产品携带的细菌数量。另一方面,在一些生产操作中,人员与物料直接接触,导致直接污染产品,都影响产品灭菌前微生物污染水平。生产操作人员应具备基本的微生物知识,养成良好的卫生习惯,按照洁净环境的要求正确穿戴防护服;设备设计、工艺设计及生产操作设计应尽可能减少人员和生产操作引起污染的风险。正常生产之后操作人员定期对手部进行清洁消毒,质量部按照洁净区手和物体表面细菌计数检测规程进行日常监测,检测结果应符合要求。
2.2.1.5灭菌前产品的初始污染菌的控制标准
对灭菌前包装好的VSD的初始污染菌设定警戒限、纠偏限以及合格限(即行动限),警戒限不宜设置过低(如低于合格限的10%)以避免过于敏感、频繁警报;也不宜过高(如高于合格限的50%)以避免失去警告的意义,本评估报告以随机三批产品灭菌前的生物负载为数据源,采用标准差法进行数据分析,分析结果分别如表2所示。
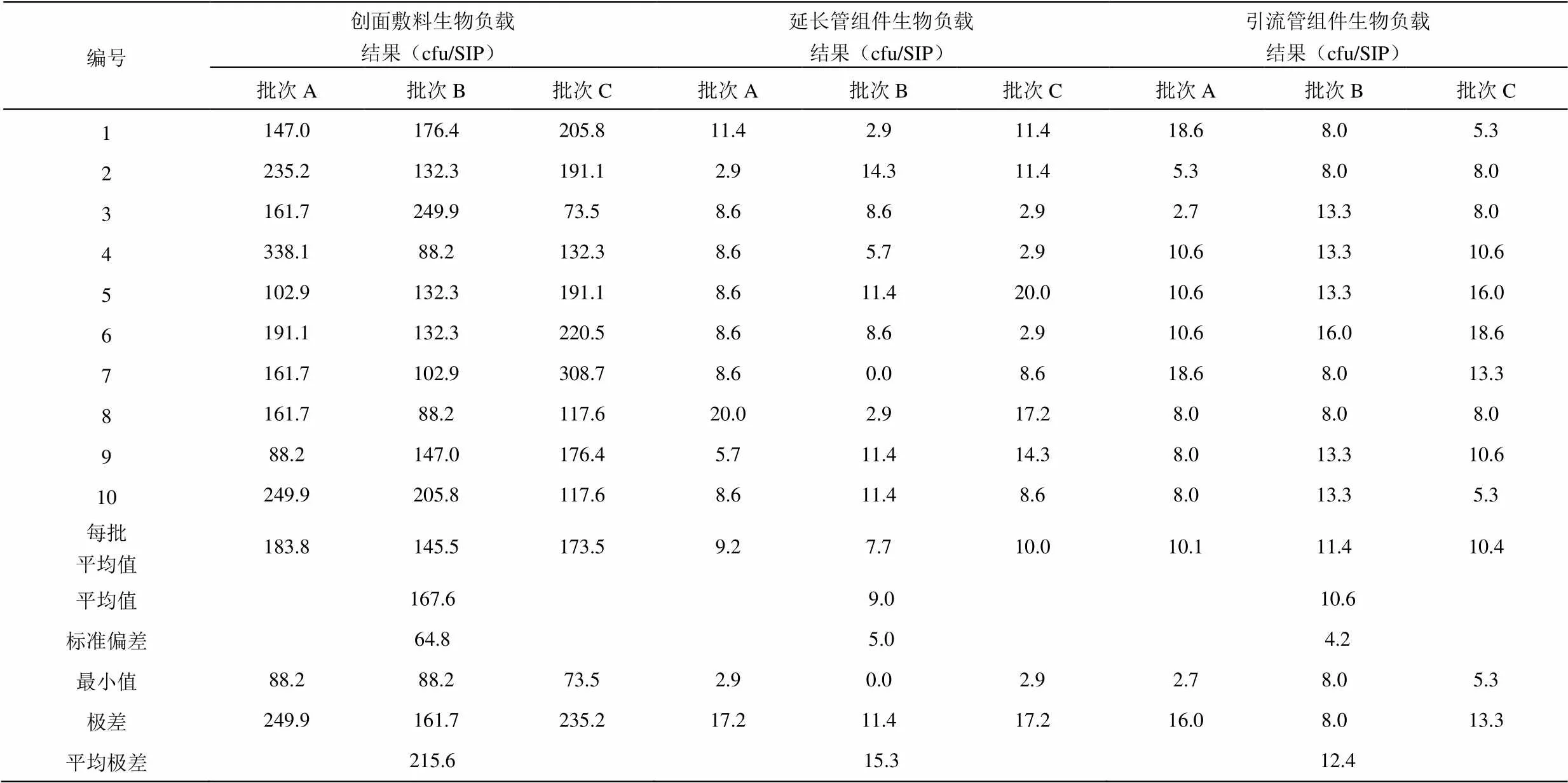
表2 3批VSD生物负载结果
一般情况下警戒限和纠偏限的计算方法为平均值加上标准偏差的倍数,通常为2或3。各部件的计算结果如下:
(1)创面敷料。
警戒限=平均值+2×标准偏差=167.6+2×64.8=297.2 cfu/SIP =297.2/60≈5 cfu/g
纠偏限=平均值+3×标准偏差=167.6+3×64.8=362 cfu/SIP =362/60≈6 cfu/g
合格限=10 cfu/g
通过标准差法计算得出警戒限为297.2 cfu/SIP,纠偏限为362 cfu/SIP,本次灭菌计量设定使用的VSD中的创面敷料规格为300 mm×200 mm×30 mm,重量约60 g,是技术要求中创面敷料最大的规格,因创面敷料的规格比较多,大小体积不一样,重量也不一样,故评定标准限度定为以每克微生物的限度比较合理,即警戒限为5 cfu/g,纠偏限为6 cfu/g,最初的合格限为10 cfu/g,警戒限是合格限的50%,符合设计要求。
(2)延长管组件。
警戒限=平均值+2×标准偏差=9.0+2×5.0=19 cfu/SIP
纠偏限=平均值+3×标准偏差=9.0+3×5.0=24 cfu/SIP
合格限=50 cfu/SIP
通过标准差法计算得出警戒限为19 cfu/SIP,纠偏限为24 cfu/SIP,最初的合格限定为50 cfu/SIP,警戒限是合格限的38%,符合设计要求。
(3)引流管组件。
警戒限=平均值+2×标准偏差=10.6+2×4.2=19 cfu/SIP
纠偏限=平均值+3×标准偏差=10.6+3×4.2=23 cfu/SIP
合格限=50 cfu/SIP
通过标准差法计算得出警戒限为19 cfu/SIP,纠偏限为23 cfu/SIP,最初的合格限定为50 cfu/SIP,警戒限是合格限的38%,符合设计要求。
需要注意的是,在正常生产之后,应定期对产品族、验证方法以及相关警戒限和纠偏限进行评审并形成记录,以确保其持续适用性。
2.2.2创面敷料细菌内毒素的污染评价与控制策略
控制了产品微生物污染,就控制了细菌内毒素的污染。由于创面敷料厂家提供的供货状态是内包完成后的状态,故对供应商要求其生产工艺中应有降低、控制微生物污染和细菌内毒素污染的措施,并且原材料敷料出厂时应对其进行全性能检测,其中包括初始污染菌以及细菌内毒素检测,结果应符合要求。来料之后本厂对其初始污染菌进行检测监控,之后在产品灭菌前再做一次VSD各组件的初始污染菌检测,以验证其生物负载在可控范围之内,灭菌之后的成品检验时,对VSD中的创面敷料进行细菌内毒素的检测,检测结果均应符合限度<20 EU/件的要求[11]。
3 验证
鉴于微生物污染菌种类的多样性,无法通过有限的试验获取生产体系的可靠性的充分证据,但毕竟验证能提供有利的证据和线索,证明一般情况下的生产体系的可靠性。验证分为初始污染菌检测验证、生产工艺验证和灭菌工艺验证三部分。
3.1 初始污染菌检测验证
通过检测VSD原材料和灭菌前初始污染菌的结果可知,初始污染菌检测结果均符合要求,并且数据相对稳定,相差不大,说明VSD原材料储存一定时间内也不会大量繁殖,威胁产品质量。
3.2 生产工艺验证
针对以上分析的风险因素,连续生产三批VSD产品,在灭菌前对其分别进行生物负载的检测,灭菌后对同一个灭菌批产品进行无菌检测以及对三批产品进行细菌内毒素检测,结果均符合要求,通过对今后检测结果的数据分析,能获取更多的信息,获得更大的质量保证。
3.3 灭菌工艺验证
产品无菌的最终保障是灭菌工艺。本公司的VSD产品采用电子束辐射的方式进行灭菌,在辐照灭菌前是基于ISO11737-1医疗器械灭菌微生物学方法的第1部分“产品上微生物群落的测定标准[12]”要求,在验证过程中首先是对三个连续独立批次的VSD进行分别随机抽取5套进行校正因子的测试,目的是对该产品的生物负载测试方法的有效性和重复性进行确认,得出修正系数,用以补偿洗脱效率。同时从这三个独立连续的生产批中分别随机抽取10套样品进行生物负载的检测,分别计算出样品中各组件的平均初始污染菌数量,如表2所示,结果显示,创面敷料、延长管组件、引流管组件和的初始污染菌平均值分别为167.6 cfu/SIP、9.0 cfu/SIP、10.6 cfu/SIP。
根据GB18280.2医疗保健产品灭菌辐射第2部分:建立灭菌计量标准中VDmax25方法[13],确定了VSD产品的辐照灭菌验证剂量为8.8±10% KGy。随机抽取10套VSD产品,用验证剂量进行辐照灭菌,并对辐照之后的产品进行无菌检查,结果显示10套样品的无菌试验结果不超过1个阳性,符合《中国药典》中无菌检查[14]标准的要求。
验证后,并采用25.0 KGy辐照剂量对VSD产品进行辐照加工,经无菌检验结果显示辐照后的产品无菌检验无一件为阳性结果,符合《中国药典》无菌检查标准的要求。在本测试条件下,证实了25.0 KGy 可确定为VSD产品在电子束辐照灭菌过程中最低灭菌计量,此剂量提供的灭菌保证水平(SAL)为10-6。
4 结论
控制产品的无菌保证和细菌内毒素质量风险以确保使用者的安全性为目标。VSD产品的生产过程并等待灭菌的工艺过程存在微生物过量繁殖潜在风险,并威胁产品的无菌保证和细菌内毒素质量。该风险需要通过有针对性的设备和工艺设计、质量管理策略等全面的风险控制方法得以控制。通过上述分析和提供的证据,可以得出以下结论:
(1)包括工艺设备和工艺流程和质量控制策略在内的整个生产体系是根据消除、控制上述风险设计并建设的。
(2)所有已知的风险都采取了针对性的措施。
(3)已建立了以控制、监测灭菌前微生物污染量为核心的质量控制策略,能及时发现偶发的灭菌前VSD异常微生物污染。
(4)产品的灭菌工艺、生产工艺和清洁验证工艺等都经过了验证,最终灭菌能有效杀灭灭菌前存在的微生物。
(5)对产品无菌保证的概念和要求具有深入的了解,有能力控制各种偶发的微生物污染风险,保证有质量风险的产品不流入市场。产品的无菌保证和细菌内毒素质量风险是完全可以控制的。
[1]YY/T 1737-2020. 医疗器械生物负载控制水平的分析方法[S]. 北京: 中国标准出版社,2020.
[2]何随梅,黄磊. 浅谈雪莲注射液的无菌保证与质量风险控制点[J]. 科技创新与应用,2014(23): 57.
[3]GB∕T 19973.1-2015. 医疗器械的灭菌微生物学方法第1部分:产品上微生物总数的测定[S]. 北京: 中国标准出版社,2015.
[4]王文庆,张步增,吴平. 手术单、手术衣和洁净服生物负载水平测定方法介绍[J]. 中国医疗器械杂志,2014, 38(2): 138-140.
[5]柯月娇,宋洪涛,张勇,等. 医院制剂室纯化水设备的验证[J]. 山西医药杂志,2020(15): 2066-2068.
[6]李明俊,莫善军. 高效处理污染物应急技术示范工程自动控制系统开发及应用[J]. 自动化应用,2017(8): 19-21.
[7] YY/T 0316-2016. 医疗器械-风险管理对医疗器械的应用[S]. 北京: 中国标准出版社,2016.
[8] 丁芬. 小容量注射剂无菌保证控制措施(最终灭菌)[J]. 科学技术创新,2018(19): 55-56.
[9] YY0033-2000. 无菌医疗器具生产管理规范[S]. 北京: 中国标准出版社,2000.
[10] GB50457-2019. 医药工业洁净厂房设计规范[S]. 北京: 中国标准出版社,2019.
[11] YY/T0618-2017. 医疗器械细菌内毒素试验方法常规监控与跳批检验[S]. 北京: 中国标准出版社,2017.
[12] ISO 11737-1: 2018 Sterilization of health care products Microbiological methods Part 1: Determination of a population of microorganisms on products[S]. Geneva: International Standard Organization, 2018.
[13] GB 18280.2-2015. 医疗保健产品灭菌辐射第2部分:建立灭菌计量[S]. 北京: 中国标准出版社,2015.
[14] 国家药典委员会.中华人民共和国药典2020版四部[S]. 北京: 中国医药科技出版社,2020.
Sterility Assurance and Bacterial Endotoxin Quality Risk Assessment of Disposable Negative Pressure Drainage Wound Care Material Package
This paper evaluates and analyzes the sterility assurance and the quality risk of bacterial endotoxin of the disposable negative pressure drainage wound care material package (VSD), and evaluates the actual risk control level of the product by analyzing the risk factors causing the initial bacterial / biological load pollution of the product before sterilization, evaluating the risk factors and taking certain risk reduction control measures. By analyzing the data and formulating reasonable control basis, the risk is controlled within an acceptable range, which proves that the sterility assurance of the product and the quality risk of bacterial endotoxin can be completely controlled.
disposable negative pressure drainage wound care material package; sterility assurance; bacterial endotoxin; initial contaminating bacteria; bioburden
TH77; R472
A
1008-1151(2022)05-0049-04
2022-02-10
李发家(1986-),男,江苏盐城人,苏州凯瑞斯生物科技有限公司工程师,从事医疗器械质量检测工作。
猜你喜欢
现代畜牧科技(2021年8期)2021-10-13
猪业科学(2021年3期)2021-05-21
心肺血管病杂志(2020年5期)2021-01-14
小哥白尼(军事科学)(2018年9期)2018-12-08
小哥白尼(军事科学)(2018年8期)2018-09-12
小哥白尼(军事科学)(2018年6期)2018-09-10
小哥白尼(军事科学)(2018年1期)2018-05-25
现代园艺(2018年3期)2018-02-10
中华老年口腔医学杂志(2016年1期)2017-01-15
湖南中医药大学学报(2016年1期)2016-12-01
