
An efficient single atom catalysts Os/P3C sheet for ammonia borane dehydrogenation
2022-07-11 03:39ChozhengHeQunZhngJinrongHuoLingFu
Chinese Chemical Letters 2022年6期
Chozheng He,Qun Zhng,Jinrong Huo,b,∗,Ling Fu
a Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices,School of Materials Science and Chemical Engineering,Institute of Environment and Energy Catalysis,Xi’an Technological University,Xi’an 710021,China
b School of Sciences,Xi’an Technological University,Xi’an 710021,China
c College of Resources and Environmental Engineering,Tianshui Normal University,Tianshui 741001,China
Keywords:Ammonia borane Two-dimensional materials Dehydrogenation Single atom catalyst Microkinetic model
ABSTRACT Ammonia borane (NH3BH3,AB) has been considered to be a promising chemical hydrogen storage material.Based on density functional theory,a series of transition metal atoms supported P3C (P3C_O) sheet is systematically investigated to screen out the most promising catalyst for dehydrogenation of AB.The results indicate that the Os/P3C and Os/P3C_O could be an efficient single atom catalyst (SACs) and the stepwise reaction pathway with free energy barrier of 2.07 and 1.54 eV respectively.Remarkably,the rate constant further quantitatively confirmed the real situation of the first step of dehydrogenation of AB on the Os/P3C and Os/P3C_O substrates.We found that kf1 at 400 K is equivalent to kf2 at 800 K,which greatly improves the temperature of the first step of AB dehydrogenation on P3C_O.We hope this work can provide a promising method for the design of catalysts for AB dehydrogenation reactions on the surface of two-dimensional materials (2D).
In recent years,the rapid increased energy demands urged the development of alternative energy sources,which are clean and renewable [1].Hydrogen is considered as one of the best candidates to satisfy the increasing demand for an efficient and clean energy carrier because hydrogen possess higher gravimetric energy density than petroleum (120 kJ/g for hydrogenvs.44 kJ/g for petroleum) and with only water as by-product [2].However,the efficient storage [3]and production of hydrogen are still two key issues in the “hydrogen economy" [4–6];.Under the continuous exploration of predecessors,it has been demonstrated that using solid media,such as sorbent materials [7–11](activated carbon,nanotubes,carbon,metal-organic frameworks,etc.) and hydrides (metal hydrides,complex hydrides,chemical hydrides,etc.)[12–14],is the safest and most effective way to store hydrogen.Among the new hydrogen storage materials,various complex hydrides,ammonia borane (NH3BH3,AB),appears to be a suitable hydrogen source and is attracting more and more interest in the field of solid-state hydrogen storage because of its abnormally high hydrogen content of 19.6 wt% and well-behaved stability under ambient conditions.Hydrogen can be released from ABviathermolysis or catalysis in various solvents [15].Al-Kukhunet al.[16].also reported the cyclization mechanism of AB low temperature dehydrogenation.Luo and Ohno [17]reported an intramolecular stepwise dehydrogenation process catalyzed by Cp2Ti in which N-H activation precedes B-H activation.However,there are some serious drawbacks which need to be overcome in order to make it suitable for practical on-board application: (1) The relatively high dehydrogenation temperature (>100°C) and low hydrogen release rate [18,19];(2) The detailed theoretical mechanism of AB releasing hydrogen in different substrates is not well understood and further studies are required [16];(3) The by-product borazine can hinder the practical application of AB because of its toxicity [20].
To overcome the above drawbacks and make the AB can be dehydrogenated under mild conditions.a number of approaches have been developed recently,including heating ammonia borane pyrolysis,acid catalysts [21–23],metal complex catalysts [24,25],metal particle and transition metal [11,15,26].Noted that the introduction of a catalytic amount of platinum (∼2 mol%) to a solution of AB in 2-methoxyethyl ether (0.02–0.03 mol/L) results in accelerating hydrogen evolution at room temperature [27]and the RuP2[28,29]nanohybrid also can be as an efficient bifunctional catalyst for hydrolysis of AB [30].Pt-based catalysts,such as small Pt nanoparticles supported on porous chromium terephthalate (MIL-101) [31],and carbon nanotubes (CNT) [32,33],exhibit superior catalytic activity for hydrolysis of AB.Transition non-noble metals such as Cu,Ni and Co [34–36]are also widely studied to improve the use efficiency of precious metals.Unfortunately,most catalysts are relatively expensive metals [37,38]or nanoparticles (such as Rh[39],Ir [40]and Os [41]) or suffer from instability under the reaction conditions [42,43].Searching for new types of catalysts is still demanding.An alternative way is to maximize the utilization of noble metal by downsizing the size of nanoparticles even to single atoms on designed substrates [44].Due to its excellent activity,selectivity and stability,single-atom catalysts (SACs) have become eternal themes in both academia and industry [45–47].Wuet al.[48].reported that Pt SACs are supported on the surface of graphene oxide as effective catalysts for the hydrolysis of AB to hydrogen.Penget al.[49].proposed that a Rh SACs supported on oxygen-rich nitrogen-doped carbon nanosheets produced highefficiency activity for AB hydrogen production.Indeed,the wide range of metal-hydrogen bond polarization present in these compounds,combined with the interaction of the metal and the charge of the AB molecule achieves the purpose of activation [50],which offers an extremely versatile platform of efficient AB dehydrogenation catalysts.
After Sunet al.[51]found that phosphene-graphene hybrid materials exhibit high performance,more and more graphene hybrid materials have been experimentally and theoretically demonstrated.Recently,a new P/C composite material (PC6) has been reported by Jianget al.[43,52],and this analogue of graphene is designed as a substrate with supported transition metals (TMs),which exhibited excellent catalytic activity.Similarly,Zhaoet al.[53].predicted a new buckled hexagonal P/C composite material(P3C),which have intrinsic metallicity and excellent thermal stability.Lin Long and his colleagues used density functional theory(DFT) calculations to reveal that when P3C is used as a catalyst substrate [54],SACs exhibit excellent catalytic activity for electrocatalytic reactions.Therefore,this new two-dimensional material may become a highly active catalytic substrate.
In this paper,we evaluated the potential of TMs supported P3C(P3C_O) sheet (TM/P3C and TM/P3C_O) for AB dehydrogenation reactions.A number of TMs were considered as the supported metals to screen out promising SACs [55]for AB dehydrogenation.Then,the adsorption and activation mechanisms of AB on the SACs surface was also revealed by the analysis of charge differential density(CDD),density of states (DOS) and crystal orbital Hamilton population (COHP).And the possible reaction pathways for AB dehydrogenation are also provided [56].On the other hand,rate constants in kinetics further quantitatively confirmed the real situation of the first step of dehydrogenation of AB.Therefore,our investigations may provide a rational route to design and evaluate the efficient catalysts for AB dehydrogenation reactions.
All the calculations are carried out with the ViennaAb InitioSimulation Package (VASP) [57]based on the density functional theory (DFT) [58].The Perdew Burke and Ernzerhof (PBE) functional within generalized gradient approximation (GGA) was used to describe the electronic exchange-correlation interaction [59,60].The projector augmented-wave (PAW) method was applied to illustrate the interaction of ion-electrons [61].The cutoff energy for the plane-wave basis is set to 400 eV.For structural elaxation and electron self-consistent calculation,the convergence criteria for energy and force are set to 10−4eV and 0.02 eV/˚A,respectively.The Gamma Scheme of 2×3×1 is also employed for the geometric optimization and 6×7×1 k-points were applied to the electronic structure calculation.The Bader charge analysis [62]was used to compute the charge transfer.At the same time,the zero-point energy (ZPE) correction [63]for the total energy was adopted.Device Studio [64]program provides several functions for performing visualization,modeling,and simulation.Transition states (TS)were searched by climbing image nudged elastic band method (CINEB) [65]and further confirmed by vibrational frequency analysis.The calculation simulation process is performed on P3C surface structure,as shown in Fig.S1 (Supporting information).The lattice vectors area=10.69 ˚A,b=12.35 ˚A,andc=15.49 ˚A.To avoid artificial interactions,a vacuum of 15 ˚A was added in the Z direction[66,67].The dehydrogenation mechanisms of AB reaction catalyzed reactions all proceed on the surface [68,69].
The 2D material P3C is obtained by calculation and prediction due to the high performance of the phosphorene-graphene hybrid material [70,71].As presented in Fig.S1a,the basic building blocks of P3C monolayers P6and P4C2rings and it owes a C2/m symmetry.We investigated the electron structure of 2D P3C by calculating the band structure and partial density of states (PDOS) (Fig.S1b).As can be seen,there are some bands crossing the Fermi level,which means it is a good conductor.2D P3C exhibits intrinsic metallic features and the high conductivities and its high thermodynamic stability was identified through Ab Initio Molecular Dynamics simulation and Phonon spectra according to the report by Zhaoet al.[72].Then,six possible adsorption sites (Bpp,Bpc,Hpp,Hpc,Tp,Tc) on the P3C monolayer are considered to confirm the energetically most favorable anchoring site for the considered TM(TMs=Fe,Co,Ni,Zr,Nb,Mo,Ru,W,Os,Zr and Pt) atoms as shown in Fig.S1a.And the most stable geometry structures of TMs/P3C sheet are shown in Fig.S2 (Supporting information).Further,the binding energy (Eb) of TM atoms on P3C monolayer is calculated to verify the stability of SACs according to the formula:Eb=EP3C-M−EP3C−EM,in which M represents different single atoms.Ebrepresents the Binding energy of M atom,EP3C-M,EP3CandEMrepresent the total energy of M adsorbed on the 2D P3C plane,2D P3C surfaces,and single atoms in bulk metals.The largeEbin Table S1(Supporting information) implies that there is a strong interaction between TMs and P3C monolayer and TM/P3C is a kind of promising and feasible SACs.
Further,charges are redistributed due to the interactions between the supported TMs and the P3C substrate.For example,some of the TMs (Os,Pt and Ir) shown by bader charge analysis have obvious electron transfer from the P3C sheet to the TMs [73],and the other is the opposite.Interestingly,it has been reported that due to the interaction between electrons,the charged TM may activate the adsorbed molecules [74,75],which are expected to adsorb and activate AB and promote the dehydrogenation reaction of AB.Therefore,TMs/P3C tablets may provide a promising method to promote the dehydrogenation of AB under mild conditions.
To estimate and design the most promising SACs based on P3C substrate,we have implemented a rational screening strategy,including the adsorption of AB molecules on the SACs according to the B-terminal and the N-terminal adsorption respectively.The corresponding structures are shown in Fig.S3 (Supporting information,B-terminal) and Fig.S4 (Supporting information,N-terminal).Remarkably,through structural optimization,we found that some of the AB molecules catalyzed by TMs/P3C (Fe/P3C,Ni/P3C,Zr/P3C and Mo/P3C) can only be stabilized on the substrate through Bterminal adsorption.In order to further determine the best adsorption position,we calculated the adsorption energy of the AB molecules at the B-terminal and the N-terminal.The adsorption energies of AB on the TMs/P3C were calculated by the following equation is defined as:Ead_AB=Etot−EP3C-M−EAB.WhereEtotrepresents the energy of the AB adsorbed on TMs/P3C.EP3C-MandEABare the calculated total energy of TMs/P3C and single AB,respectively.Specifically,if the adsorption energyEad_ABis a positive value,it means that the adsorption of the AB molecule on the SACs is endothermic.On the contrary,ifEad_ABis a negative value,it means that the adsorption is a spontaneous process.As shown in Fig.S5 (Supporting information),the black represents the adsorption energy of the N-terminal,and the red represents the adsorption energy of the B-terminal.The higher adsorption energy negative represents the more stable the AB molecule is adsorbed on TMs/P3C.We found that the B-terminal adsorption of the AB molecule is more stable in most cases (except for W/P3C).Therefore,all the subsequent results are developed around the Bterminal adsorption of AB (Fig.1a).And all the detailed results during the screening progress are listed in Fig.1b (pink line chart).First of all,the absorption of AB molecules is an important criterion for the subsequent AB activation and dehydrogenation reaction.Thus,the calculated adsorption energy of AB molecule on surface as following: Co/P3C (−0.922 eV)
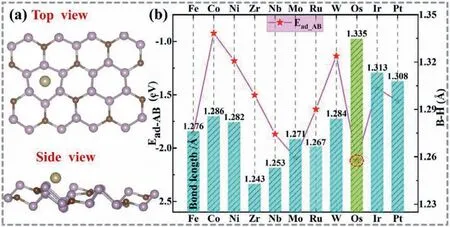
Fig.1.(a) Top and side view of Os adsorbed on P3C sheet (Os/P3C).(b) The adsorption energy of AB (Ead_AB) for different sites and corresponding bond length of B-HII (AB)on the Os/P3C.
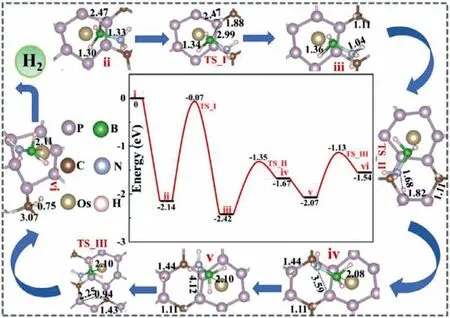
Fig.2.The complete free energy (ΔG) correction diagram and related structure diagrams for the dehydrogenation of AB molecules on the surface of Os/P3C.
In the present study,we found that the reactant AB on the surface of Os/P3C can be dehydrogenated by stepwise pathway: in its first half,a B-H bond is broken to form an intermediate characterized by an H atom adsorbed at the top of C atomsviaa H-adsorbed transition state;in the second half,the adsorbed B-H bond is broken and the two adsorbed H atoms form a hydrogen moleculeviaa transition state of the H movement.More interestingly,we found that O doping on the P3C surface (Os/P3C_O) will greatly reduce the adsorption energy of AB,thereby weakening the B-H bond energy in the AB molecule,making the dehydrogenation pathway of the AB reaction easier.Selected optimized structure for the stationary points along the pathways is given in Figs.2 and 3.The relative energy values are the Gibbs free energy (ΔG) (the surface energy of 2D P3C (P3C_O) is selected as 0 level energy) except for specially mentioned,the corresponding correction data are listed in Tables S3–S6 (Supporting information).As shown in Fig.2,the structure of the stepwise reaction pathway shows that one B-H bond and NH bond in the adsorbed AB are gradually elongated,and then the H proton migrates on the surface to form a hydrogen molecule,and the remaining BH2NH2species adsorbed on the Os atom.Firstly,AB molecules are chemically adsorbed on Os/P3C,and the adsorbed B-HIIbond changes from 1.219 ˚A to 1.335 ˚A.The loosening of the bonds facilitates the dissociation of the first H atom in the AB dehydrogenation pathway.As shown in the structure of Fig.2,the elongated B-HIIbond undergoes a transition state TS_I (false frequency is 1328.08 cm−1),and the bond length has changed from the original 1.335 ˚A to 2.993 ˚A,which is almost completely broken.The energy barrier of this process is 2.07 eV,which is diffi-cult to occur at room temperature,but it is worth noting that the heat released by AB molecules adsorbed on Os/P3C is 2.14 eV,so we firmly believe that the heat released by AB adsorption can provide the heat required for self-reaction.The shed H atoms move to the C atoms on the P3C surface to form an intermediate iii with a C-H bond of 1.11 ˚A.Noted that the B-HIbond has been elongated to 1.36 ˚A,so there are two possible ways of evolution,namely Bterminal continuous dehydrogenation (Fig.S8 in Supporting information) and the stepwise reaction pathway (Fig.2).B-HIIfracture undergoes a transition state TSII (false frequency is 1315.22 cm−1)absorbs heat of 1.4 eV,and the energy barrier crossed is 2.61 eV.Both kinetics and thermodynamics are unfavorable.Thence,our main consideration is the stepwise reaction pathway of the N-HVIpath.
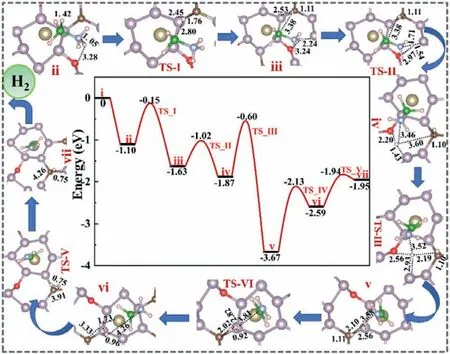
Fig.3.The complete free energy (ΔG) correction diagram and related structure diagrams for the dehydrogenation of AB molecules on the surface of Os/P3C_O.
In this pathway,the N-HVIbond slightly activated by the surface undergoes a transition state TS_II (false frequency is 1300.91 cm−1)with an energy barrier of 1.07 eV.Simultaneously,the HVIatom moves to the surface to form a P-H bond with a bond length of 1.44 ˚A,∗NH2BH2(∗indicates adsorbate) gradually begins to move away from Os/P3C to form an intermediate iv,and then∗NH2BH2undergoes an energy-free flattening process and leaves slightly Os/P3C surface.After that,two H atoms on the surface cross the transition state with an energy barrier of 0.94 eV to form TS_III(false frequency is 727.27 cm−1).In this process,the H atoms on the two surfaces approach each other to form a bond length of 0.94 ˚A of H2molecules,while C-HIIand P-HVIare elongated to 1.43 and 2.25 ˚A,respectively.The generated H2molecules are far away from the surface and accompanied by a shortening of the HH bond (0.75 ˚A),which is highly consistent with the bond length of normal H2molecules (0.75 ˚A).This process absorbs heat of 0.64 eV.The present results indicate that the dehydrogenation of the AB molecule on the Os/P3C surface are feasible due to the entire reaction is exothermic and has a low energy barrier.
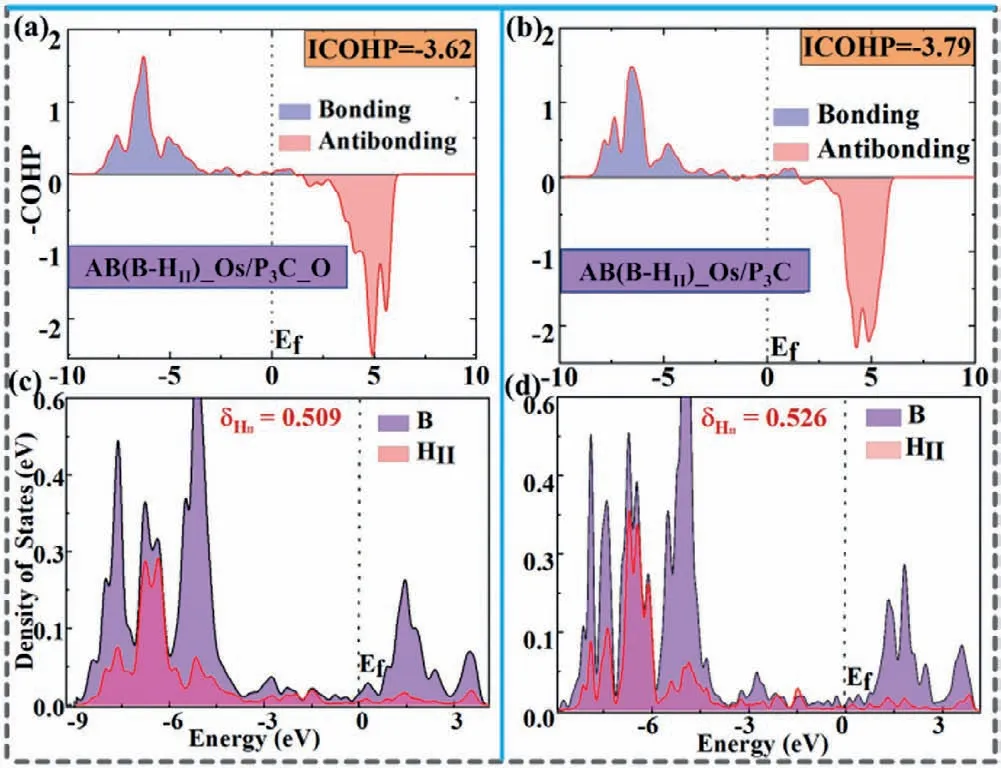
Fig.4.(a,b) The crystal orbital Hamiltonian population (COHP) of the B-HII bond in the AB molecule adsorbed on Os/P3C_O and Os/P3C,respectively.The bonding and anti-bonding states in ICOHP are represented by light blue and light red.(c,d) The calculated corresponding partial density of states (PDOS).
We replaced the C atom near the Os adsorption site with an O atom to form an oxidized P3C_O substrate [76].In order to systematically evaluate the stability of the substrate,we further evaluated the stability of P3C_O through AIMD simulation [77].As shown in Fig.S9 (Supporting information),the structure maintains well at 800 K.The geometric structure is still not significantly deformed,indicating that P3C_O has high thermodynamic stability.We further carried out the MD simulation of 18 ps,the energy and temperature oscillated near the equilibrium state,and the structure of P3C_O remained good.This shows that P3C_O can exist stably at the actual reaction temperature.As shown in Fig.S7,the P3C surface doped with O stretched the B-HIIbond length to 1.457 ˚A,and the N-HVI bond length to 1.053 ˚A.Based on the degree of activation of the AB molecule B-HIIand N-HVIby the Os/P3C_O substrate,we studied the same dehydrogenation pathway.As shown in Fig.3,the AB molecule is adsorbed on Os/P3C_O with an adsorption energy of −1.10 eV forms an adsorbed substance ii,undergoes a transition state TS-I (false frequency is 1371.89 cm−1),which elongates the bond length of B-HIIto 2.45 ˚A,and the HIIatoms gradually transfer to the C atoms on the surface,forming an intermediate state iii with C-HIIof 1.11 ˚A.After that,N-HVIbegan to be elongated and experienced TS-II (false frequency is 468.03 cm−1),and the dropped HVIcontinued to approach the surface,forming O-HVIof 2.20 ˚A and P-HVIof 1.43 ˚A.In the intermediate iv,the HVIon the surface undergoes TS-III (false frequency is 1084.53 cm−1)and approaches HII.Simultaneously,the∗NH2BH2adsorbate starts to move away from the surface,forming a vertical structure of v with the surface of the substrate.After that,the HVIatoms on the surface pass TS-IV (false frequency is 663.99 cm−1) continues to move towards the HIIatom,forming an intermediate vi with HVI-HIIof 0.96 ˚A above the P atom near Os.Finally,the formed HVI-HIIundergoes TS-V (false frequency is 61.41 cm−1) and begins to move away from the surface and the vii with HVI-HIIof 0.75 ˚A is formed,and the distance between the H2molecules and the surface reaches 4.26 ˚A,completely detaching from the surface.In addition,∗NH2BH2can continue the dehydrogenation reaction or leave the surface to form by-products.These results indicate that the stepwise reaction pathway for AB dehydrogenation on the Os/P3C_O is energetically favorable.
The mechanism of AB activation is also evaluated theoretically,firstly,we investigated the initial adsorption configuration and electronic structure of AB adsorbed on Os/P3C and Os/P3C_O.As shown in Table S2 (Supporting information),doping with O atoms makes the adsorption energy of AB changed from −2.14 eV to −1.10 eV.Interestingly,the bond length of B-HIIhas grown from 1.335 ˚A to 1.417 ˚A,and N-HVI(1.053 ˚A).Compared with the original B-HII(1.217 ˚A) and N-HVI(1.032 ˚A),the AB molecule is fully activated.To figure out the nature of the interaction between supported Os atom and AB,CDD is analyzed to illustrate the interaction mechanism.It can be found from Figs.S7b and c that the charge transfer and redistribution can be observed around the AB molecule and supported Os atom,respectively.To deeply understand the activation process,ICOHP and DOS of the B-HIIbond in the first dehydrogenation reaction of AB on Os/P3C and Os/P3C_O are calculated in Figs.4a and b.The decrease of ICOHP values of B-HIIbond (from −3.79 to −3.62) confirms that the strength of BHIIbond of Os/P3C_O is obviously weakened.In order to deep insights into the interaction between atoms in bonding,we analyzed the interaction of the B-HIIbond by plotting PDOS.The size of the overlapping peak area of PDOS indicates the strength of the bond between atoms.It can be seen from Figs.4c and d that the integral value (0.526) of the Os/P3C surface is greater than Os/P3C_O(0.509),this result once again explains the nature of the low energy barrier of the Os/P3C_O substrate adsorption of AB in the first step of dehydrogenation reaction.

Table 1 The relationship between the equilibrium constant (kfi) of the reaction and the reaction temperature (T), kf1 and kf2 represent the first step dehydrogenation equilibrium constant of AB on Os/P3C and Os/P3C_O,respectively.
Rate constant [78]was performed to further verify confirmed quantitative analysis of Os/P3C and Os/P3C_O activated AB at different temperatures.The relationship between rate constants (kf i)and reaction temperature (T) is shown in the following formula[79]:

whereviis the pre-exponential factor,andEactis the ZPE-corrected activation energy.Within harmonic transition state theory,we can calculate the pre-exponential factor (νi) of each reaction pathway using the following definition:

whereandare the vibrational frequencies at the initial state and the vibrational frequencies at the transition state (excluding the imaginary one).
From the above formula,we can deduce that the relationship between the first step dehydrogenation rate constant on Os/P3C and Os/P3C_O:

whererf1andrf2are the ratio of the first step dehydrogenation rate of Os/P3C and Os/P3C_O;θABis the coverage of AB on the surface.kis the ratio of dimensionlessrf1torf2.
Applying the same rough rule of thumb [78],the dehydrogenation energy barriers of AB in the first step of Os/P3C and Os/P3C_O are 2.07 and 0.95 eV,their reaction temperatures are 800 K and 400 K,respectively.Based on the DFT results and microkinetic model,we have calculatedkf1andkf2of 300–850 K by Eqs.1 and 2.From the data listed in Table 1,we can find that thekf1(kf2) is increased with increasing temperature due to increase it is conducive to the increase in the average kinetic energy of the molecules,the increase in the number of activated molecules and the effective collision frequency.Obviously,with the increase of temperature,the increase ofkf1andkf2and the value ofkis getting smaller and smaller,which indicates that there is a certain limit in the reaction of AB on Os/P3C and Os/P3C_O,and the sensitivity to temperature keep getting smaller.And it is worth noting that thekf1at 400 K and thekf2at 800 K are in the same order of magnitude.This not only proves the accuracy of the empirical formula [75],but also shows that the P3C surface doped with O greatly reduces the temperature of the reaction so that the reaction occurs under mild conditions.
In our work,a DFT calculation was performed to design and evaluate the catalytic activity of TMs/P3C sheet for AB dehydrogenation.There is a strong interaction between TMs and P3C substrate,which can alter the local geometric and electronic structure and promote AB dehydrogenation reaction.According to our screening criteria,the Os/P3C can obviously activate the AB due to the electron exchange interactions.And Os/P3C (Os/P3C_O) sheet is considered as a very promising electrocatalysts for AB,which needs to suffer from a moderate Gibbs free energy barrier of 2.07 and 1.54 eV respectively.Simultaneously,the microscopic kinetic model further quantitatively confirmed the real situation of the first step of dehydrogenation of AB on the Os/P3C and Os/P3C_O substrates.We found thatkf1at 400 K is equivalent tokf2at 800 K,which greatly reduced temperature of the first step of AB dehydrogenation on Os/P3C_O.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No.21603109),the Henan Joint Fund of the National Natural Science Foundation of China (No.U1404216),the Special Fund of Tianshui Normal University,China (No.CXJ2020-08),and the Scientific Research Program Funded by Shaanxi Provincial Education Department (No.20JK0676).Jinrong Huo was partially supported by the postgraduate research opportunities program of HZWTECH (HZWTECH-PROP).Thanks for the National Supercomputing Center in Zhengzhou.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.02.055.
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria