
Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
2022-07-11 03:38ZeleChenJirongChenJunXun
Chinese Chemical Letters 2022年6期
Zele Chen,Jirong Chen,Jun Xun,∗
a Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials and Key Laboratory of Functional Inorganic Materials of Anhui Province,College of Chemistry &Chemical Engineering,Anhui University,Hefei 230601,China
b CCNU-uOttawa Joint Research Centre,College of Chemistry,Central China Normal University,Wuhan 430079,China
Organic molecules containinggem–difluoromethylene unit are one of the most important classes of compounds that have various valuable applications ranging from drug discovery to material science [1].Not surprisingly,numerous strategies have been developed towards the synthesis of these target organofluorine compounds.Within this research area,direct C–F bond cleavage of cheap and readily accessible polyfluorinated compounds represents one of the most potentially powerful methods to access diversegem–difluoromethylene skeletons.Compared with the well developed ionic types of C–F bond cleavage processes [2],defluorinative functionalization CF3groupsviathe formation of fluoroalkyl radicals is relatively overlooked.Recent synthetic efforts using photochemical strategy mainly focused on the Ar–CF3systems,in which single-electron reductive generation of arene radical anion followed by a fluoride anion elimination was proposed as the key step to form the fluoroalkyl radicals [3–9].However,the requirement of electron-deficient substituents (generally-CN group)on trifluoromethylarenes and the relatively low synthetic value of the formed benzylicgem–difluoromethylene products limited the method for further application.Wang and Houk recently expanded the reaction substrates to selective C-F bond cleavage of trifluoroacetamides and acetatesviaa boryl radical enabled spin-center shift (SCS) strategy [10].Despite high efficiency,the harsh reaction conditions (stoichiometric DTBP,high reaction temperature)still left space for the exploration of alternative green and mild reaction systems.
In seeking a solution to overcome the afore-mentioned challenges,Molander and co-workers uncovered a mild photochemical defluorinative alkylation of trifluoroacetates and trifluoroacetamides by a rational combination of visible light-promoted hydrogen atom transfer (HAT) and single electron transfer (SET) process (Scheme 1A) [11].From the mechanistic insights,the suc cess of Molander’s reaction can be attributed to the following aspects,(1) the suitable bond dissociation enthalpy (BDE) of the reaction components,e.g.,alkyl thiols (S−H BDE=87−89 kcal/mol),the newly formed C−H bond in product 3 (97−99 kcal/mol) and the formyl C−H bond (87 kcal/mol),which can guarantee the efficiency of the designed polarity matched HAT process;(2) the very negative reduction potential of photo-generated CO2•−species(Ev=−2.2 V) serves as strong single electron reductant to activate a broad array of challenging substrates,including trifluoroacetates or-acetamides 1 (Ev=−2.0 V) to afford radical anion 5;(3) compared with Wang and Houk’s work with the formation neutral radical intermediate for SCS process,the charged species radical anion 5 undergoes much faster defluorinative SCS to accelerate the formation ofgem–difluoro radical 6.
The designed reaction occurred under the irradiation of 390 nm Kessil Lamp with 20 mol% of diary ketone 4 as photoredox catalyst and cyclohexyl thiol as HAT co-catalyst,which offers both functional group compatibility (good tolerance of alcohols,amides,amines,anhydrides,epoxides,boronate esters,and electron-rich heterocycles) and broad substrate scope (Scheme 1B).A wide variety of olefins,such as unactivated terminal alkenes,internal alkenes,enamines,enamides,and enol ethers all participated well in this transformation to give the corresponding defluorinative functionalization products with good yields and regioselectivities.More importantly,the reaction could be applied to the structural modification of alkene-containing natural products (3-I–3-IV),demonstrating the applicability of the current method in the construction of complex molecular architectures.The authors also revealed that styrene derivatives were not suitable for this photochemical defluorinative functionalization protocol.
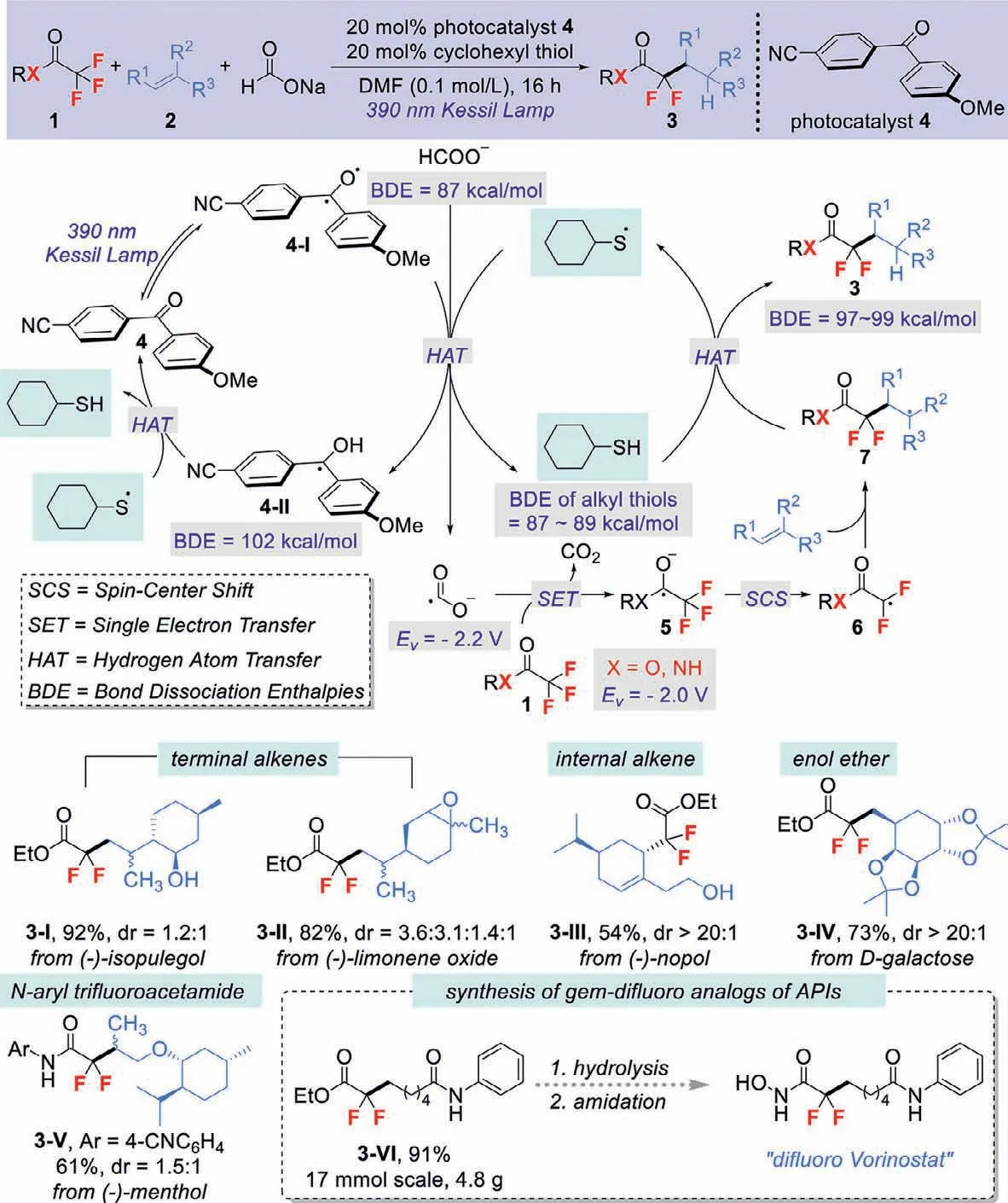
Scheme 1.gem-Difluoromethylene unit synthesis through defluorinative functionalization.
Importantly,this strategy constitutes a robust and general approach for the defluorinative functionalization of trifluoroacetamides (3-V as an example).The authors found that the addition of 20 mol% Zn(OTf)2can facilitate the SET reduction step by lowering the reduction potential ofN-aryltrifluoroacetamides by 0.5 V.More significantly,most of the remaining trifluoroacetamide start materials could be recovered through simple crystallization before chromatography.
Another notable feature of this photochemical defluorinative functionalization process is that it can easily be scaled up without the need for specialized equipment or irradiation source.In one example,the transformation was performed on a 50 mmol scale in the presence of only 1.0 mol% diary ketone catalyst.The product 3-VI,precursor ofgem–difluoro analog of APIs “difluoro Vorinostat”could be obtained in 91% isolated yield at 17 mmol scales,showing the value of this technology in medicinal chemistry.
In conclusion,the elegant work of Molander group on defluorinative functionalization of trifluorinated carbonyls demonstrates valuable advancements to the field of photochemical C–F bond cleavage.The radical chain mechanism allows for direct generation of high reactive CO2•−acting as a potent SET reductant for activation of typically redox inert trifluoroacetates and trifluoroacetamides,thereby providing an exciting opportunity for accessing value-addedgem–difluoromethylene-containing architectures in good yields and regioselectivities.The successful structural modification of alkene-containing natural products and concise synthesis ofgem–difluoro analogs of APIs further renders the approach attractive and valuable.Note that,the two successive defluorinative alkylations of CF3groups to make monofluoro products was not achieved and the reactions of trifluoroacetamides were only limited toN-(4-cyanophenyl)-trifluoroacetamide are two current limitations of the method.
Following this publication,two similar photochemical defluorinative functionalization polyfluorinated carbonyls were also independently reported by Glorius group (polyfluorinated aliphatic amides and esters) [12]and Chatterjee group (trifluoromethyl ketones) [13].Notably,Shang and co-workers realized the photocatalytic defluoroalkylation or hydrodefluorination of trifluoroacetamides,trifluoroacetates,and trifluoromethyl (hetero)arenes using cheap and easily accessibleo-phosphinophenolate as photoredox catalyst [14].All of those nice contributions further proved the importance and significance of this fascinating field.
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria
- Recent progress on the smart membranes based on two-dimensional materials