
Multiple exosome RNA analysis methods for lung cancer diagnosis through integrated on-chip microfluidic system
2022-07-11 03:39YunxingLuZhoduoTongZhenhuWuXioyuJinLinZhouShihuiQiuChunjieShenHoYinHongjuMo
Chinese Chemical Letters 2022年6期
Yunxing Lu,Zhoduo Tong,Zhenhu Wu,Xioyu Jin,Lin Zhou,Shihui Qiu,Chunjie Shen,Ho Yin,Hongju Mo,∗
a State Key Laboratory of Transducer Technology,Shanghai Institute of Microsystem and Information Technology,Chinese Academy of Sciences,Shanghai 200050,China
b Center of Materials Science and Optoelectronics Engineering,University of Chinese Academy of Sciences,Beijing 100049,China
Keywords:Exosome Integrated processing RNA analysis Microfluidics Droplet digital PCR
ABSTRACT Exosomes are now raising focus as a prospective biomarker for cancer diagnostics and prognosis owing to its unique bio-origin and composition.Exosomes take part in cellular communication and receptor mediation and transfer their cargos (e.g.,proteins,mRNA and DNA).Quantitative analysis of tumor-related nucleic acid mutations can be a potential method to cancer diagnosis and prognosis in early stages.Here we present an integrated microfluidic system for exosome on-chip isolation and lung cancer RNA analysis through droplet digital PCR (ddPCR).Gradient dilution experiments show great linearity over a large concentration range with R2=0.9998.Utilizing the system,four cell lines and two mutation targets were parallelly detected for mutation analysis.The experiments demonstrated mutation heterogeneity and the results were agree with cell researches.These results proved our integrated microfluidic system as a promising means for early cancer diagnosis and prognosis in the era of liquid biopsy.
Lung cancer is one of the malignant tumors among worldwide which resulted in maximum number of cancer death [1].Because of the inconspicuous symptom in early stages and frequentlyoccurred metastasis and recurrence,lung cancer would be diagnosed in middle and advanced stages [2,3].As an accurate and noninvasive method,liquid biopsy [4–7]offers a promising future for cancer diagnosis.
Exosomes are one of the biotargets in liquid biopsy [8–10].Exosomes are a kind of nanovesicles (∼30–200 nm in diameter)that play a critical role in intercellular communication and “cargo”transformation [11].Derived by eukaryotic cells into bioliquids,exosomes have several outstanding superiorities,like high abundance(∼1010particles in 1 mL plasma),high stability in bioliquids and easy acquisition.Secreted by parental cells,especially tumor cells,exosomes can be the “trucks” of proteins and nucleic acids “cargos”[12–14].In addition,hereditary information of parental cells can be read out through analyzing the nucleic acid components.Exosomal nucleic acids [15]include mRNA,lncRNA,miRNA and some DNA.Nevertheless,on account of complicacy of sample preparation,developing convenient and effective methods for analyzing exosome nucleic acids is in urgent need.
Real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR) is one of the RNA detection methods [16],where expression of target gene is detected by reading out the fluorescence intensity of whole reaction-tube [17,18],therefore,detection biases may occur and rare mutation may be ignored in background noise.Digital polymerase chain reaction (dPCR)is a new detection method that achieved single-molecule detection [19–21],which is free from standard curves and provides a far more accurate method to rare RNA detection [22,23].In our recent researches,dPCR has proven itself as a powerful tool against exosomal cancer-derived lncRNA [24]and SARS-CoV-2 RNA [25].
In this study,we developed an integrated multiple fluorescence droplet digital PCR (ddPCR) microfluidic system for on-chip exosome isolation and RNA analysis (Fig.1),which is sensitive,unbiased,high-throughput and inexpensive.Utilizing the system,exosome samples were firstly on-chip isolated in the processing chamber,mingled with lysis buffer and form reaction mixture.After that,the mixture was transformed through the microtubule into the liquid entrance and converged with mineral oil at the standard droplet structure.During the exosome processing and droplets generation,no off-chip operation was involved,thus liquid contamination was avoided.∼25 μL reaction mixture was formed and up to 100,000,50μm-diameter droplets were generated and contained into reaction chamber,which is much higher than commercial chips (30,000–50,000) and the dead volume is much lower (∼5% compared to 30%).Beside,a polymethyl methacrylate(PMMA) packing structure was assembled for liquid stabilizing and maintain the holistic system.Dilution testing showed the great stability of our system (R2=0.9998),beside,the multiple mutation analysis of 2 genes in 4 cell lines were consistent with cell researches [8].These results indicate our system to be a promising solution for exosomal RNA analysis.
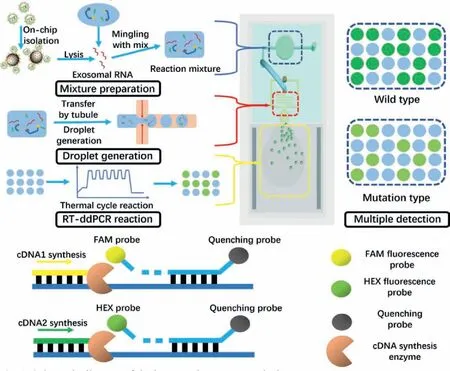
Fig.1.Schematic diagram of the integrated exosome analysis system.
To be specific,the microfluidic system is consisted of four main sections.The exosome on-chip processing chamber,droplet generation structure,the droplet reaction chamber and the PMMA packing structure.The exosome processing chamber and droplet generation structure were fabricated by polydimethylsiloxane (PDMS)and connected with a microtubule.The droplet reaction chamber was composed by two-layered glass with a 50μm gap where droplets were maintained for single-layered reaction.Additionally,the PMMA packing structure was fabricated by machine tool for liquid transformation.The processing chamber was a through-hole punched by puncture needle,7 mm in diameter,4 mm high,allowing the volume over 120 μL for high flux reaction.As a comparison,typical microfluidic chamber fabricated by pattern transformation,was usually less than 10 μL.Therefore,our system was capable to high volume reaction with a larger concentration range.
For droplet generation structure,standard cross-shaped droplet generation structure was designed.During our experiments,exosome reaction mix prepared in the chamber was transferred through micro-tubule into liquid entrance,free from pipette,and eliminated the potential risk of contamination.After generated,droplets were flowed into the large-volume (up to 25 μL) doublelayered glass chamber for RT-ddPCR reaction.Aiming at sufficient liquid transformation,a PMMA packing structure was designed and manufactured to maintain and stabilize the injection pressure of reaction phase.The structure packed the microfluidic system around and covered the processing chamber,avoiding pressure from decreasing by divulging.
The microfluidic system was fabricated according to standard process for prototyping of microfluidic systems in PDMS.The pattern mold of the exosome procession platform was manufactured by standard soft lithography.To be specific,the pattern portrayed by CAD software was firstly printed onto the chromium photo mask,followed by projecting to a silicon wafer photoetching.After that,deep reactive ion etching (deep RIE) techniques and sulfuric acid rinsing were utilized for structure etching.Subsequently,the silicon mold was treated with trichloro-(1H,1H,2H,2Hperfluorooctyl) silane (Sigma Aldrich,St.Louis,MO,USA) vapor overnight and ready for chip molding.
For microfluidic system fabrication,the PDMS (Sylgard 184 resin),cross-linker,and Triton X-100 (Sigma-Aldrich,St.Louis,MO,USA) were mixed at the ratio of 10:1:0.05 (w/w/w) and degassed under the vacuum condition.The mixture was then poured onto the silicon mold to form the patterned microfluidic structure.Baked at 70°C for 50 min,the PDMS structure was carefully exfoliated and punched the inlet and outlet.Lastly,the microfluidic structure was bonded to a clean glass through air plasma treatment.Then microtubule inserted to connect the outlet of processing chamber and liquid entrance.The droplet reaction chamber was assembled by two pieces of glass slides with a 50μm thick double-sided adhesive (DSA).Between the micro channel and the cubage,an interval was designed (named “venting gap”) for air bubbles elimination caused by pressure fluctuations.PMMA packing structure was depictedviaSolidworksTMand manufactured through machining workshop,then polished and assembled by screw and nut set.For system operation,the microfluidic system was packed by the PMMA structure and the processing chamber was sealed,therefore stable gas pressure for droplet generation was guaranteed.
Aiming at characterizing the scale distribution and concentration of exosomes,nanoparticle tracking analysis (NTA) (Particle Metrix,Meerbusch,Germany) was utilized under the standard protocols.To be specific,10 μL exosome samples were diluted in phosphate buffer saline (PBS) to reach the proper concentration(108–109particles/mL).During the detection,particles were radiated by laser and captured and tracked by a CCD camera.The measurements were conducted at 20±3°C.Then the size distribution and concentration were determined by the NTA 3.2 Analytical Software Suite and shown in Fig.2a.A unimodal peak around 120 nm could be observed,which was in accordance with published researches.
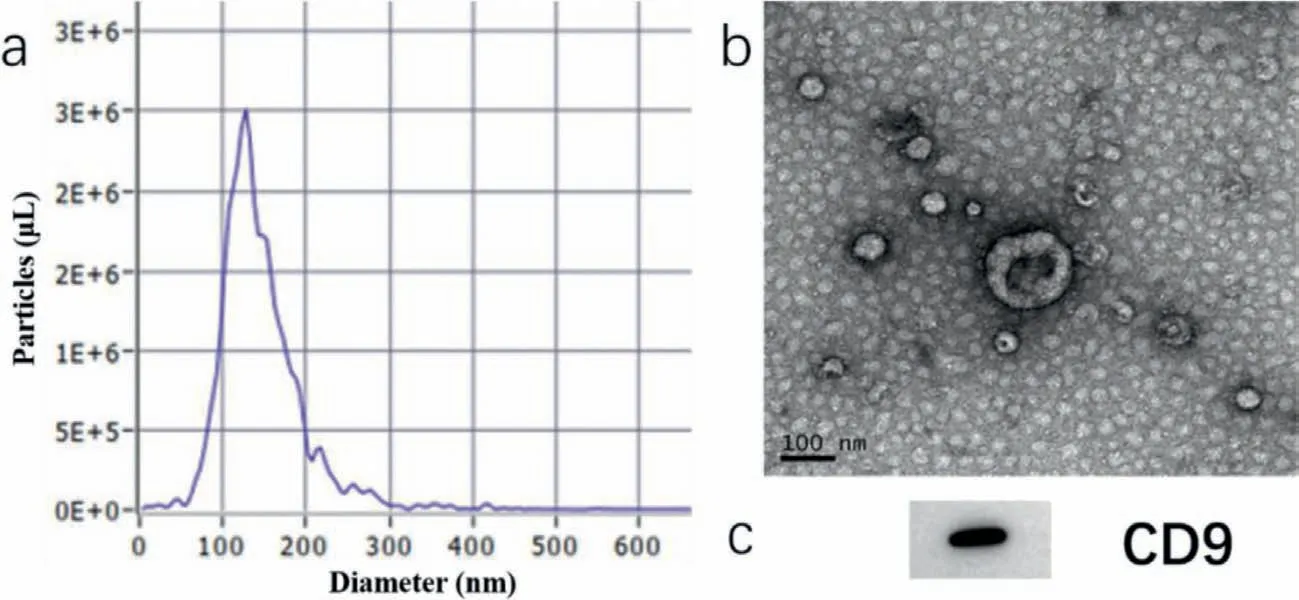
Fig.2.Exosome characterization through NTA (a),TEM (b) and western blot (c),scale bar 100 nm.
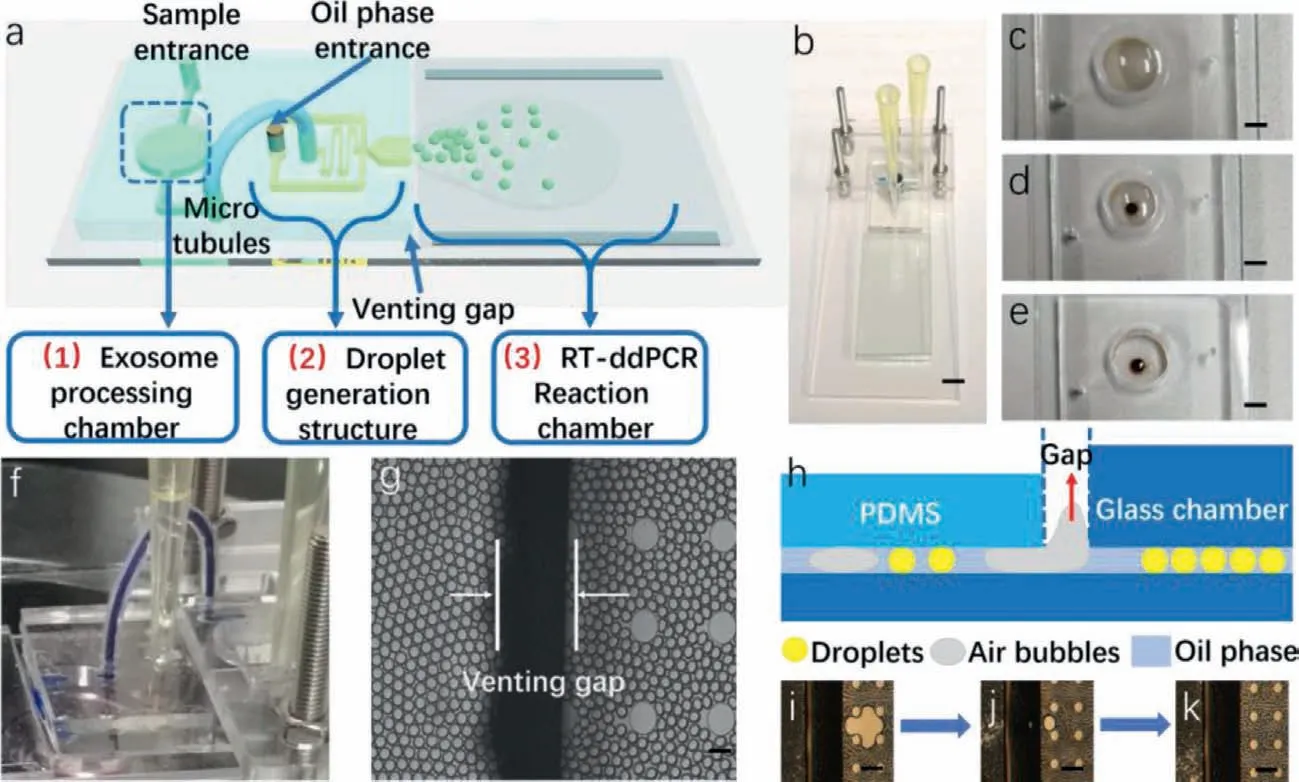
Fig.3.Four main parts of the microfluidic system,(a) schematic diagram of the whole system,(b) prototype of the system,scale bar 2 mm,(c–e) exosome isolation processing in the chamber,scale bar 1 mm,(f) microtubule-based liquid transformation,(g) micrograph of the venting gap,scale bar 100μm,(h–k) schematic diagram and feature of the venting gap for air bubbles elimination,scale bar 200μm.
For transmission electron microscopy (TEM),exosomes were dripped onto a bronze parafilm by pipette and washed by PBS sequentially,then quiescence for 10 min to drain exosomes and added a drop of 2% paraformaldehyde,dyed in 2% uranyl acetate at room temperature for exosome counterstain,followed by embedded in 0.13% methyl cellulose and 0.4% uranyl acetate for 10 min,and then imaged by microscopic observation.Fig.2b showed the morphology of exosomes,and typical saucer shape could be observed.
For western blotting,exosome sample was then treated with loading buffer at the ratio of 4:1 to obtain protein lysate.After being boiled at 95°C for 5 min,the product then cooled down and conducted with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).Followed by PVDF transformation,the lysate was incubated with anti-CD9 at 1:5000 and bound with horseradish peroxidase (HRP) secondary antibody.At last,the immunoreactive band was visualized under imaging system.The result was shown in Fig.2c.
The whole exosome processing could be observed owing to the transparent chip (Figs.3a and b).In the exosome isolation process,the immune magnetic beads were dispersed in the exosome samples as shown in Fig.3c.After isolation,a magnet was placed under the chamber and the beads were collected at the center of the bottom (Fig.3d) and the supernatant was excluded (Fig.3e).Then,the lysis buffer and reaction mix were added in order to form the final mix.
Following that,the exosome chamber was sealed and the chip was packed with the PMMA structure.The PMMA structure was designed to stablize the high gap pressure in the droplet generation.After assembly,the reaction mix was successfully transformed into the liquid entrance in Fig.3f (replaced by blue ink for observation convenience).
During the usual droplet generation,it is worthy to note that air bubbles would be generated from pressure fluctuation.Bubbles would expand and shrink during the thermal cycle process,and cause droplets fusion and affect the subsequent analysis.In our system,this problem could be solved.A double-layered reaction chamber was placed below the droplet structure and forming a venting gap shown in Fig.3g.When bubbles accidently generated,they would rise out from the gap due to the floatage difference between liquid and air (Fig.3h).Figs.3i–k showed the whole processing of air bubbles elimination.
DdPCR is one kind of absolute quantification detection method.Different from qPCR,which measures through Ct value,ddPCR counts the number of positive droplets and total droplets and calculates the copy number by Poisson distribution theory.Thus,it is of importance to set threshold to distinguish positive droplets from negative droplets.
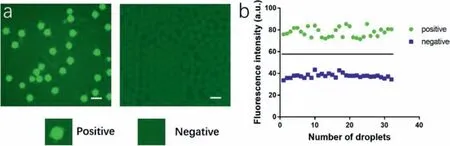
Fig.4.Droplets distinguishing.(a) Micrograph of droplets after thermal cycle reaction (left) and negative control (right),inserts are representative positive and negative droplets,scale bar 100μm,(b) gray values of total droplets groups after data classification.
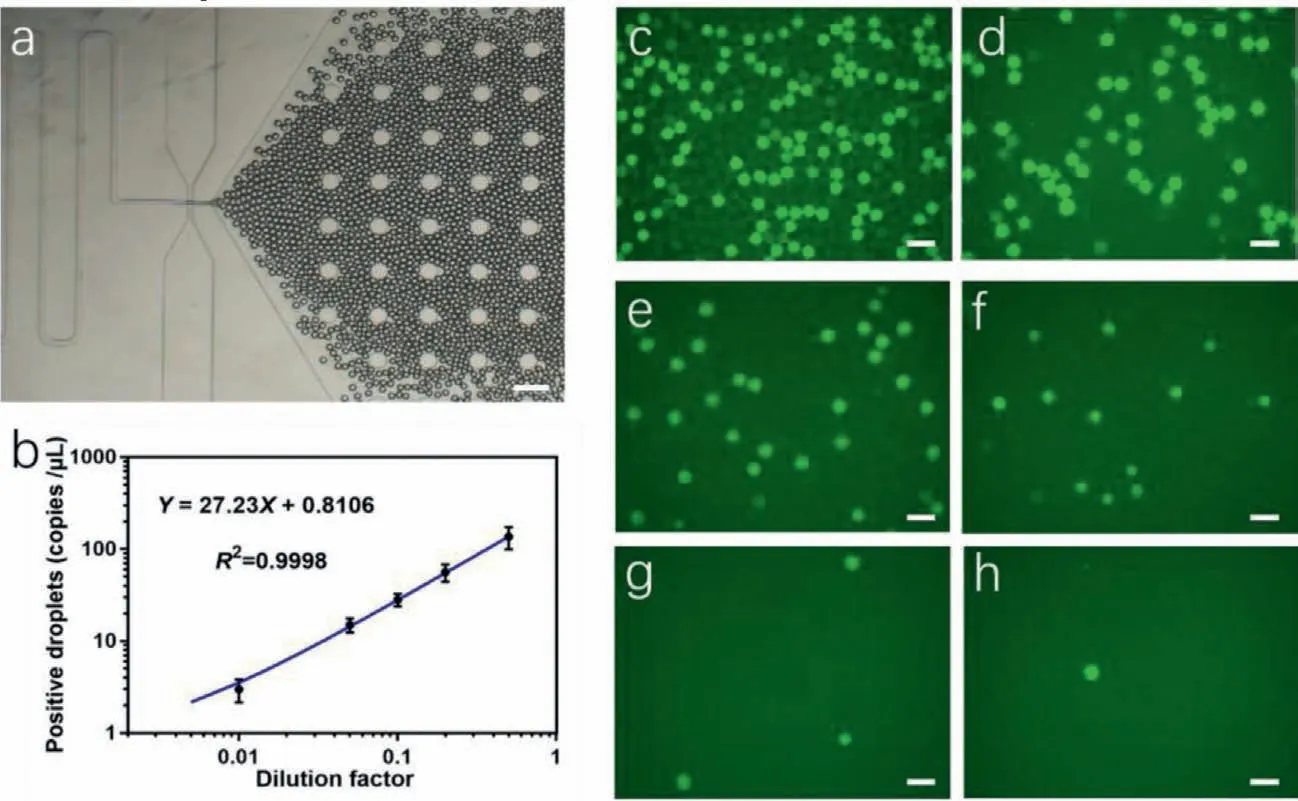
Fig.5.System performance examination.(a) Micrograph of droplets generation,scale bar 100μm,(b) linear regression fitting of the dilution exosome samples,(c–h) micrographs of droplets with a series of dilution factors of sample exosomes,scale bar 100μm.
Compared to qPCR,ddPCR is self-contrasting,where both positive and negative units are analyzed together and we can distinguish positive signals by calculating fluorescence intensity of droplets.Image pro plus 6.0 was utilized to measure the gray values of total droplets (including positive and negative droplets)and data were classified into two groups.Fig.4a left showed the droplets with reaction mixture after reaction and Fig.4a right revealed the negative control (blank mixture with RNase-free water) to further avoiding false positive result.Beside,the gray values of all droplets were classified into two significant difference groups in Fig.4b,which were positive group and negative group.The mean value of positive group was 77.70 while the negative group was 37.69.Therefore,we set 57.695 as the threshold for droplet distinguishing.
Beside,we tested the linearity of copy number variation to concentration of exosome sample under gradient dilution.Linearity reflected the accuracy of the system,a better linearity means better consistency to different concentration of samples and lead to more accurate experimental results,andvice versa.
Ultracentrifuged samples from H1299 cell line supernatant were tested.Firstly,the concentration of samples was measured by NTA.Then the samples were divided into 6 groups at different dilution ratios.Following that,these samples were parallelly tested under the same operation procedure and the copy numbers were calculated and summarized.
Fig.5a showed the droplets generation process,Figs.5c–h represented the micrographs of droplets with the dilution factor at 0.5,0.2,0.1,0.05,0.01 and 0.001,respectively.Data was calculated and summarized in Fig.5b.Linear regression fitting showed linear relationship between copy number and dilution factor,withR2=0.9998,which showed great detection accuracy of our microfluidic system.Through NTA,the concentration of exosomes is measured as 3.9×107μL−1.10 μL sample was tested in the experiments and therefore,the limit of detection (LOD) is down to 3.9×105exosomes.
Mutations onEGFRandKRASare well-known in specific types of non-small cell lung cancers (NSCLCs).Identification of different mutant subtypes would benefit the cancer therapy [26,27].Therefore,to inspect the detection and distinguish ability of our system,we employed four kinds of exosomes from cancer cell lines,including H446,H1299,H1975 and A549,for on-chip exosome multiple mutation analysis.EGFR L858RandKRASgene mutations were chosen to be the detection targets in our study.
Through parallel experiments,Fig.6 exhibited the representative micro-photos of the results.Figs.6a–h wereEGFR L858Rmutant results for exosomes from H446,H1299,H1975 and A549,under carboxyfluorescein (FAM) and hexachlorofluorescein (HEX) fluorescence field,respectively,while Figs.6i–p represented theKRASmutation for same cell line exosomes.From contrast experiments it was observed that exosomes from H1975 carriedEGFR L858Rmutation,and the other three kinds of exosomes (from H446,H1299 and A549) were wild type.Beside,forKRASmutation analysis,exosomes from A549 cell lines exhibited wild type,while other three kinds of exosomes (from H446,H1299 and H1975) showed mutation type.Note that there were several mutation types forKRAS,thus in this set of experiments we concentrated on whether one kind of exosomes express mutation or not.It is proved from our researches that exosomes from various cell lines exhibited heterogeneity,and the mutation of exosomes was in accordance to their parental cells.These results revealed the potential utilities of exosomes for non-invasive diagnosis and detections.
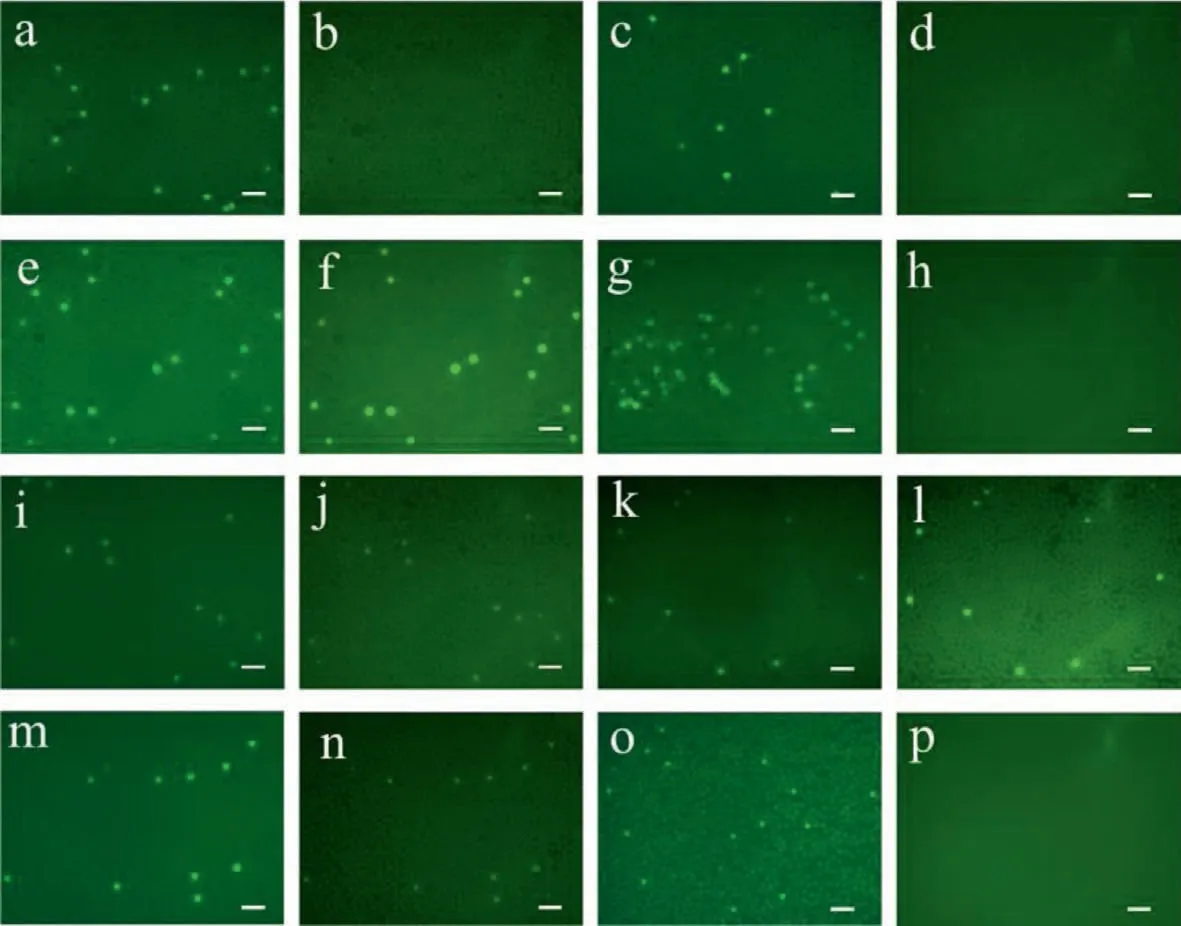
Fig.6.Parallel detections of four kinds of exosomes for multiple EGFR L858R and KRAS mutation analysis.(a-h) showed EGFR L858R mutant results for exosomes from H446,H1299,H1975 and A549,respectively,and (i-p) showed the KRAS mutation for same exosomes.Scale bar 200μm.
In this research,we present an integrated microfluidic system for exosomal RNA analysis.Exosome on-chip isolation,lysis and droplet-based digital RT-PCR reaction were accomplished through three connected functional sections,where off-chip contaminations were avoided.After the PCR thermal cycles,exosomal RNA was reacted with reaction mixture.Positive droplets exhibited significant fluorescence signal from background signal.Beside,gradient dilution parallel experiments revealed good linearity over the large concentration range,withR2=0.9998.For exosome mutation analysis,two oncogenes (EGFRandKARS) were explored in four kinds of cell lines including H446,H1299,H1975,A549 and turned out the differentiation results which was in accordance with published cell researches.
Additionally,large volume of one-stop exosome sample and reaction mixture were achieved in our system for high throughput exosome isolation and the final reaction droplets reached∼100,000.Our microfluidic chip demonstrated advantages including low-cost,high convenience,no contamination risk and disposability perspectives.By adjusting reaction mixture,it is also suitable for other gene analysis.The easily-assembled PMMA structure also facilitated sample testing.In short,our microfluidic system is prospective to exosomal RNA sensitive detection and analysis in precision diagnosis era.
Declaration of competing interest
The authors declare that they do not have any known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are grateful for the support of grants from National Natural Science Foundation of China (Nos.61971410,and 62001458),and Shanghai Sailing Program (No.20YF1457100).
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria