
Shared-probe system: An accurate,low-cost and general enzyme-assisted DNA probe system for detection of genetic mutation
2022-07-11 03:39LidRenZhihoMingWeiZhngYngweiLioXiofengTngBeiYnHuiminLvXinjinXio
Chinese Chemical Letters 2022年6期
Lid Ren,Zhiho Ming,Wei Zhng,Yngwei Lio,Xiofeng Tng,Bei Yn,Huimin Lv,Xinjin Xio,∗
a Centre of Reproductive Medicine/Family Planning Research Institute,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430030,China
b Department of Obstetrics and Gynecology,Third Hospital of Shanxi Medical University,Shanxi Bethune Hospital,Shanxi Academy of Medical Sciences;Tongji Shanxi Hospital,Tongji Medical College,Huazhong University of Science and Technology,Taiyuan 030032,China
Keywords:Nucleic acid probe Endonuclease IV Secondary structure Genomic mutation SNP detection
ABSTRACT Enzyme assisted DNA probes are powerful tools in molecular diagnostics for their simplicity,rapidity,and low detection limit.However,cost of probes,difficulty in optimization and disturbance of secondary structure hindered the wider application of enzyme assisted DNA probes.To solve the problems,we designed a new system named shared-probe system.By introducing two unlabeled single stranded DNA named Sh1 and Sh2 as the bridge between probe and the substrate,the same sequence of dually labeled probe with stable performance was shared for different mutations,thus sparing the expense and time cost on designing,synthesizing and optimizing corresponding probes.Besides,the hybridization between Sh1 and the substrate could overcome secondary structures,which guaranteed the detection of different substrates.The performance and generality of the design were tested by low abundance detection in synthetic single DNA samples and the limit of detection was 0.05% for PTENR130Q,EGFR-L858R and 0.02% for BRCA1-NM007294.3.In genomic DNA samples,the limit of detection of 0.1% can be achieved for EGFR-L858R,demonstrating the potential of clinical application in our design.
Tumor is known to result primarily from genetic mutations.Detection of genetic mutation has a significant role in molecular diagnostics of tumor.To date,researchers have developed various methods to detect mutations,such as next-generation sequencing [1–3],digital PCR [4,5],electrochemical chips [6–8],and DNA probes [9–29].Among those methods,DNA probes were widely used because of their convenience and simplicity [9–16].DNA probe-based methods without nuclease,including molecular beacons [13,14],LNA probes [12,15,16],PNA probes [30–35]and branch migration-based DNA probe [9–11,29],had the advantages of simplicity and stability,but their sensitivity was not very high.The detection limits were generally 0.5%,which was not suffi-ciently accurate to detect tumor DNA in early stage [9–16,26,28].To address this problem,one of the most common approaches employed was the coadministration of DNA probe with enzyme.Various enzyme-assisted DNA probe systems have been established[17–27,36].The utility of enzyme significantly improved the detection limit of DNA probe-based method.
However,this strategy still exhibited some shortages.Firstly,the researchers have to design and synthesize corresponding DNA probe for each mutation [9–26],which increased the cost for the need of fluorophores,quenchers and labels.Secondly,each DNA probe has to be optimized through repeated trial-and-error process due to the uncertainty of each probe’s performance [17–21]and unpredictable side-activity of enzyme [22–26].This directly caused increased cost and decreased efficiency.Thirdly,the hybridization between DNA probe and substrates may be disrupted by the secondary structures of substrates [37–39].As a result,enzymeassisted DNA probes were difficult to be applied when the genomic mutation presented in areas with higher GC content [38],which were prone to form secondary structure such as hairpin.In general,despite good detection ability,enzyme-assisted DNA probes’clinical application is substantially limited by high optimization/detection cost as well as lack of generality.Thus,there is an urgent need to develop new enzyme-assisted DNA probe systems that can overcome these disadvantages.
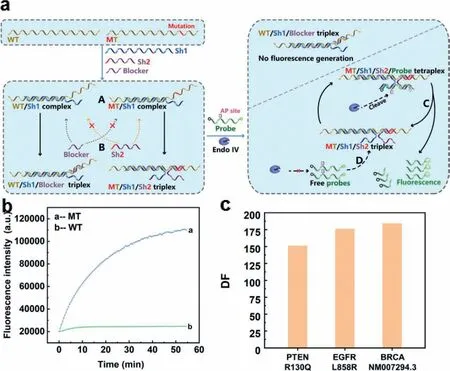
Fig.1.(a) The schematic illustration of shared-probe system.WT is wild-type substrate and MT is mutant-type substrate.Endo IV indicates Endonuclease IV.Letter “A”indicates the duplex formed by Sh1 and WT/MT to overcome possible secondary structure;letter “B” indicates the specific hybridization between Sh2/blocker and MT/WT;letter “C” indicates cleavage,release and signal generation of dually labelled probe in the tetraplex formed by MT,Sh1,Sh2 and probe;letter “D” indicates target recycling system.AP site denotes apurinic/apyrimidinic site that can be recognized and cleaved by Endo IV in double stranded DNA.(b) The fluorescence signal response of sharedprobe system towards substrates of weak secondary structure.(c) Discrimination factor of shared-probe system on 59 nt synthesized single-stranded substrates of PTENR130Q EGFRL858R/ BRCA1NM007294.3.
Herein,we designed a new enzyme-assisted DNA probe system called shared-probe system.The assay was based on Endonuclease IV (Endo IV),which has a very high affinity to double strand DNA(dsDNA) [40–44].Endo IV can recognize the apurinic/apyrimidinic(AP) site in dsDNA and degrade the single strand that contains the AP site.The oligonucleotide components and detection process were shown in Fig.1a.The shared-probe system contained four types of DNA: double labelled DNA probe with AP site,substrate to be tested (MT/WT,MT is mutant-type substrate and WT is wild-type substrate),blocker strand,and Sh1/Sh2 strands to bridge DNA probe and substrate.The Sh1/Sh2 strands consisted of 2 parts,which were probe binding region (PBR) and substrate binding region (SBR).For Sh1,its PBR is perfectly matched with an 11-nt segment of the probe at 3′ terminal,while the SBR was perfectly matched with a 25-nt segment upstream of the mutation in the substrate.For Sh2,its PBR part could hybridize with the 10 nt at the 5′ terminal of the probe,and the SBR could hybridize to the segment of substrates where the mutation lay (the mutation is marked in red in Fig.1a).The SBR of Sh2 was suggested to be 11–16 nt in length and experiment were acquired to determine the proper length for different mutations.MT and SBR of Sh2 were completely matched while WT and SBR of Sh2 had a mismatch.To have better differentiation,we introduced the blocker to compete with SBR of Sh2.The blocker was of the same length with SBR of Sh2.In contrary with Sh2,the blocker was completely complimentary with WT and had a mismatch with MT,which would mostly hybridize with WT,leaving MT free for probe.At the set temperature,Sh1 and the substrate would overcome possible secondary structures forming stable MT/Sh1 duplex and WT/Sh1 duplex (marked with “A” in Fig.1a).For WT/Sh1 duplex,the WT/Sh1 duplex and blocker would form WT/Sh1/Blocker triplex (marked by“B” in Fig.1a),which was unable to generate florescence signal.For MT/Sh1 duplex,since Sh2 was completely matched with MT,the MT/Sh1 duplex,Sh2 and probe would form MT/Sh1/Sh2/probe tetraplex.Then Endo IV would recognize and cleave AP site in the tetraplex,and the cleaved probes would dissociate from the complex to generate fluorescence signals (this process was marked by“C” in Fig.1a).Next,free probe and MT/Sh1/Sh2 triplex would form MT/Sh1/Sh2/probe tetraplex again (marked by “D” in Fig.1a) and repeat process C,generating florescence signal constantly.Experimental results in Fig.1b showed that shared-probe system was well powered to distinguish between MT and WT with a high discrimination factor (DF).The DF could be calculated by comparing the increase rates of fluorescence intensity of MT over that of WT,and the result in Fig.1c was 151,176,184,respectively for 59 nt synthesized single-stranded substrates of PTENR130Q,EGFRL858R and BRCA-NM007294.3.
It is worth noting that due to the introduction of Sh1 and Sh2,the cost of optimization and detection is substantially reduced.In Shared-Probe system,the sequence of probe is completely isolated from the sequences of substrates,which means researchers can choose a probe with the best performance for all sites of interest and synthesize probes in large amount.Further on,the optimization is much cheaper since we only need to resynthesize unlabelled Sh1 and Sh2 if necessary.Another improvement is that shared-probe system has outstanding generality by eliminating the secondary structure formed by substrates.In the system,the matching region of Sh1 and the substrate is 25 nt in length,which can hinder substrates from forming secondary structures near the detected mutations.Overall,due to these advantages,the Shared-Probe system realized the generality of double labelled DNA probe to all sites and significantly reduced the cost on both optimization and detection,which have great potential in further clinical application.
Our first priority was to select the most appropriate probe.Since the probes applied to different mutations had the same sequence in our design,the choice of probe would significantly influence the detection efficiency of the system.Here,we considered the stability as the criterion for the selection of probes.As we found in previous work [22,23,25,26],endonuclease IV had a sideactivity as an exonuclease.This activity would cause degradation of single stranded probes,generating high background signal.It was known that this unpredictable side-effect occurs on probes of some sequences while not influencing others.The side-activity is highly correlated with the sequence of AP probe.However,the reason for this correlation is currently unknown.Therefore,it is difficult to predict whether this side activity will occur or not.To find the most appropriate probe,we synthesized 8 probes (named probe-1 to probe-8,see detailed sequence in Table S1 in Supporting information) with different sequence and length and tested their stability with the existence of Endo IV.As it was explained,probes that generated less fluorescence were better in stability.The result in Fig.S1 (Supporting information) demonstrated that the best stability is obtained from probe-8,which generated little fluorescence signal.Therefore,we selected probe-8 and designed our sharedprobe system based on its sequence.
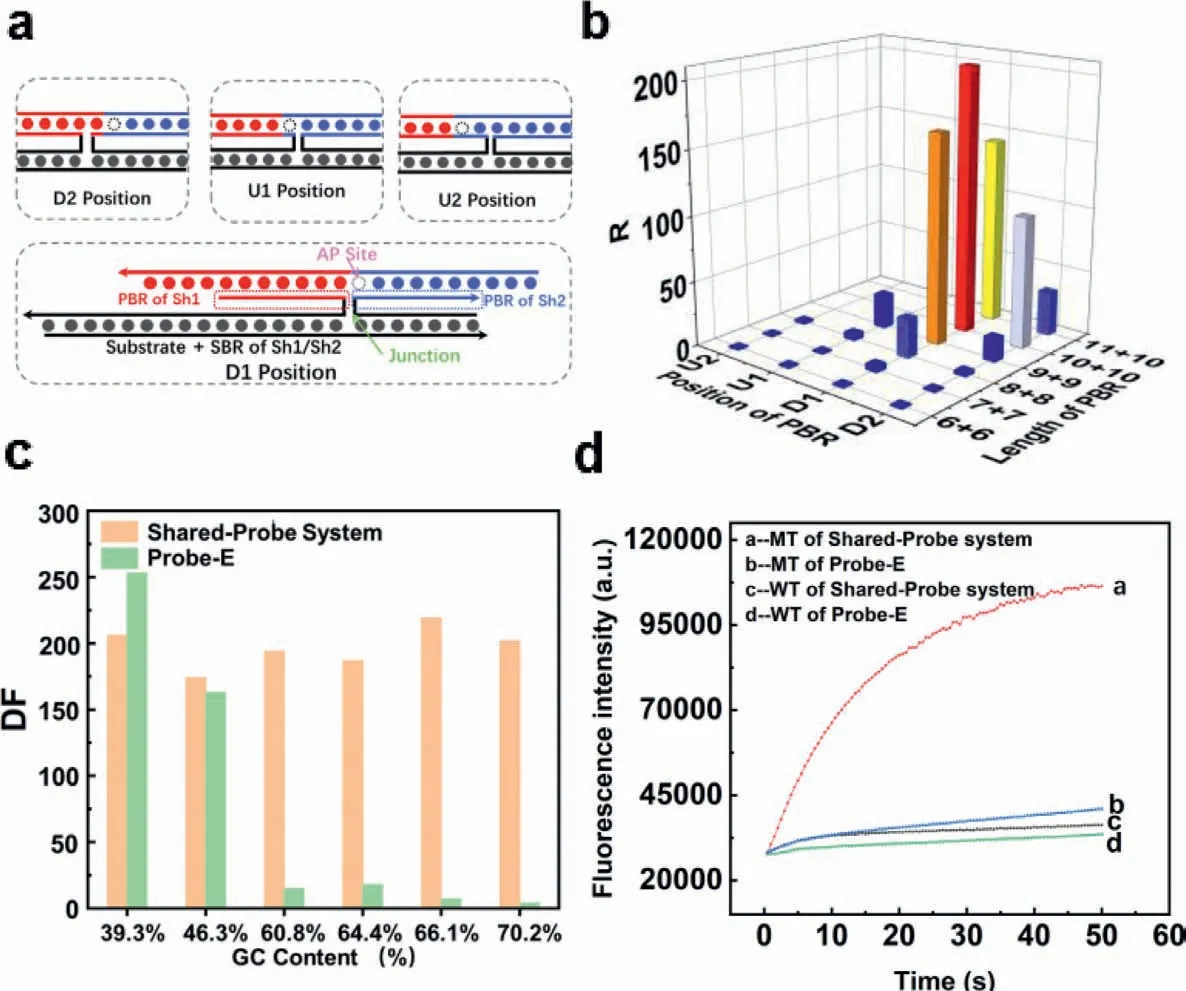
Fig.2.(a) Illustration of the tetraplex.The strand in red (left of the AP site) and blue (right of the AP site) denotes double labelled probe.The strand in totally black denotes the substrate.The strand in black and red denotes Sh1 (the red part denotes PBR while the black part denotes SBR).The strand in black and blue denotes Sh2 (the blue part denotes PBR while the black part denotes SBR).PBR were framed by dashed lines.AP denotes apurinic/apyrimidinic site that can be recognized and cleaved by Endo IV in double stranded DNA.Junction denotes the linkage between PBR and SBR of Sh1/Sh2.“U” and “D” indicated the relative position of the AP site while the number “1”and “2” indicated the distance from the AP site to the junction of PBR and SBR (substrate binding region).For example,“D1” meant the junction was at the first position downstream of the AP site.(b) R value of 19 pairs of Sh1 and Sh2.R was defined as the ratio of fluorescence increasing rate with/without substrates.The length of PBR was denoted in the form of “the length of Sh1’s PBR+the length of Sh2’s PBR”.For instance,“7+7” represented that the PBR of Sh1 and Sh2 were both 7 nt in length.(c) The differentiation factor of Shared-Probe System/Endo IV assisted probe towards 59 nt substrates of different level of GC contents.(d) The fluorescence response of Shared-Probe System/Endo IV assisted system towards the substrate of 70.2% GC content.
After the probe selection,the next step is to optimize the probe binding region (PBR,shown in Fig.2a) of Sh1 and Sh2 that will be the basis of the design of our method.The key challenge was to balance the binding of free probes and the dissociation of cleaved probes.On the one hand,the sum of length of PBR must be long enough to ensure that free probe could bind to MT/Sh1/Sh2 triplex.On the other hand,neither the PBR of Sh1 nor the PBR of Sh2 could be too long,otherwise Sh1/Sh2 might combine with the probe without the help of substrates,resulting in high back-ground signal.Furthermore,the position of the AP site also influenced recognition,cleavage and dissociation [22,26].We then designed and synthesized 19 different pairs of Sh1/Sh2 strands (named a series U-Sh and D-Sh,see detailed sequence in Table S1) by adjusting the length of PBR and the relative position of AP site in the complex.As shown in Fig.2a,the probe was 21 nt in length with the AP site at the 10th nucleotide from its 5′ terminal.“U” and “D” indicated the relative position of the AP site while the number “1”and “2” indicated the distance from the AP site to the junction of PBR and SBR.For example,“D1” meant the junction was at the first position downstream of the AP site.The length of PBR was denoted in the form of “the length of Sh1’s PBR+the length of Sh2’s PBR”.For instance,“7+7” represented that the PBR of Sh1 and Sh2 were both 7 nt in length.To make sure that the length of SBR would not influence the result,the SBR was fixed at the same length (25 nt for Sh1;20 nt for Sh2) for the 19 pairs of Sh1/Sh2.R in Fig.2b was defined as the ratio between the rate of fluorescence generated with/without the substrate added.The larger R for a pair of Sh1/Sh2 meant more fluorescence generated by the tetraplex they formed and lower back-ground signal produced without the substrate.Experimental results in Fig.2b demonstrated that the most suitable design of PBR was “D1,10+10”,which meant the junction was at the first position downstream of AP site and PBR of Sh1/Sh2 was 10 nt in length.Based on the result,we employed“D1,10+10” in our further study.
The design of PBR and SBR of Sh1 were the same for any mutation of interest,while SBR of Sh2 needed to be designed and optimized for different mutations.With the help of NUPACK,we suggested the SBR of Sh2 to be 11–16 nt in length for substrates of different sequences.To provide the most proper length,experiment was carried on for each mutation we detected in our study,and the optimized SBRs of Sh2 for PTENR130Q,EGFRL858R,BRCA1NM007294.3 were 13 nt,12 nt and 15 nt in length respectively (experiment results were shown in Figs.S2 and S3 (Supporting information)).
We have established and optimized the design principles of shared-probe system.To better demonstrate the function of shared-probe system in overcoming secondary structure of substrates,we synthesize a series of substrates with different GC contents.Based on those substrates,we compared shared-probe system and Endo IV assisted probe (probe combined to substrate directly,denoted as “probe-E” to be distinguished from the probe of shared-probe system) by their ability of differentiating MT/WT(strands are named Sc-MT,Sc-Sh1/Sh2,see detailed sequence in Table S1).We fixed the length of Sh2 at 25 nt to eliminate the secondary structure.As was shown in Fig.2c,Endo IV assisted probe had better DF (differentiation factor) when the GC content of substrates was 39.3%.However,when GC content was higher than 60%,DF of Endo IV assisted Probe decreased dramatically.To the contrary,GC content had little influence on DF of shared-probe systems.Fig.2d demonstrated the comparison of DF with and without Shared-probe system under the GC content of 70.2%.As was reported by previous literature,GC content was significantly positively correlated with the complexity of secondary structure [37,38].The experimental results above demonstrated that our strategy had good generality and could be applied to detect mutation in substrates with complex secondary structure.
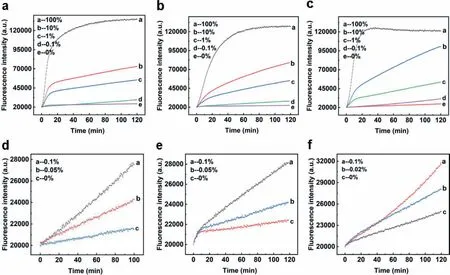
Fig.3.(a,d) Detection of low abundance PTENR130Q mutation in 59 nt synthesized DNA strands.(b,e) Detection of low abundance EGFR-L858R mutation in 59 nt synthesized DNA strands.(c,f) Detection of low abundance BRCA1-NM007294.3 mutation in 59 nt synthesized DNA strands.
To further demonstrate the generality of a detection system,its functionality on different mutations of clinical importance should be examined.We selected three common high-frequency mutation sites to be our modeling target: PTENR130Q,BRCA1-NM007294.3,and EGFR-L858R.Based on the sequence of the three target mutation sites,we synthesized corresponding 59 nt wild-type and mutant-type as substrates (named by PTEN/EGFR/BRCA,see detailed sequence in Table S1).Three corresponding Sh1/Sh2 pairs were also designed and synthesized while the same sequence of probe was used for all the three target sequences.Experimental results displayed in Fig.S3 (Supporting information) showed that our design could easily differentiate wild-type/mutant-type of the three target sequences.The DF reached 151 for PTENR130Q,176 for BRCA1-NM007294.3,and 184 for EGFR-L858R,indicating its generality and practicability (demonstrated in Fig.S4 in Supporting information).
It has been demonstrated that our design was able to overcome complex secondary structure and can be applied to multiple mutations.However,in practical application situations,mutant DNA to be detected was always at low amount [45–47].Therefore,low abundance detection on different mutations was conducted to further examine the detection ability of shared-probe system.With corresponding wild-type strands,we diluted the mutant-type strands of PTENR130Q,BRCA1-NM007294.3 and EGFR-L858R into a series of mixed substrates of different mutation abundance.Experimental results in Fig.3 showed that the limit of detection was 0.05% for PTENR130Q and EGFR-L858R,and 0.02% for BRCA1-NM007294.3,demonstrating the ability of shared-probe system inS detecting low abundance mutations.
In addition to low abundance mutation detection,we also detected the same substrates based on BRCA-NM007294.3 at different concentrations.For pure MT,our system could discriminate substrate from blank with the concentration as low as 50 pmol/L.
As for mixed substrate containing 10% of MT,100 pmol/L of substrate was successfully detected.The experiment results were demonstrated in Fig.S5 (Supporting information).
All the experiments above were conducted with synthesized DNA strands,while in clinical applications,substrates were double strands from genomic DNA.Thus,we investigated whether our design could achieve a low limit of detection in real clinical samples.The work flow was illustrated in Fig.4a.EGFR-L858R mutanttype was extracted from tissue samples of a patient with nonsmall cell lung cancer and wild-type genomic DNA was extracted from a healthy person who did not carry EGFR mutation (result of Sanger sequencing was shown in Fig.S6 (Supporting information)).We diluted mutant-type DNA by wild-type DNA into a series of different abundance.After standard PCR amplification and unparallel PCR,single stranded products were produced for detection.As illustrated in Figs.4b and c,our design reached the limit of detection as low as 0.1%,which was in line with the result using synthetic substrate.
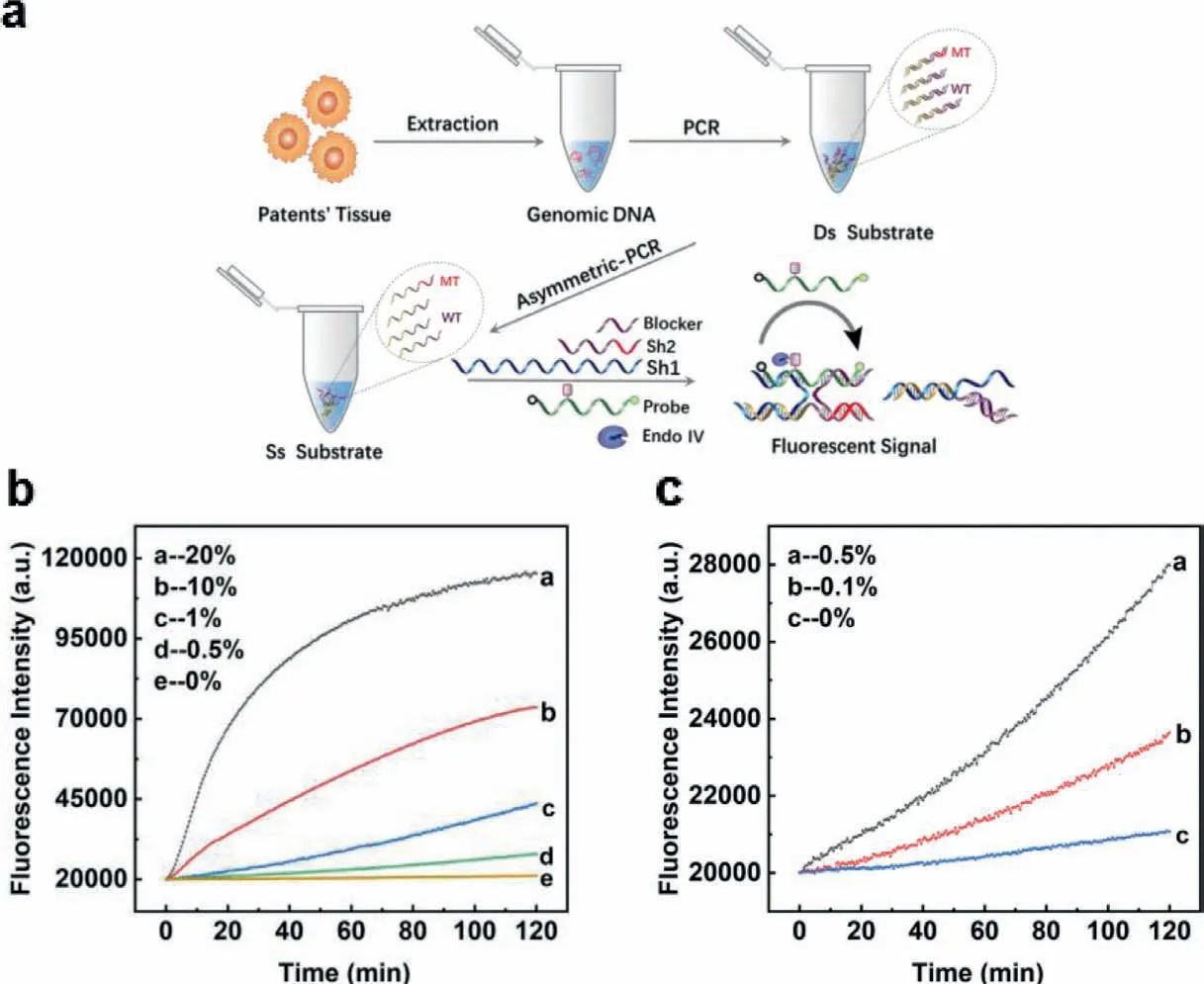
Fig.4.(a) The work flow of the whole detection process with clinical sample.(b,c) Detection of low abundance EGFR-L858R mutation in 110 nt genomic DNA sample.
In summary,we have established a mutation detection method named shared-probe system with reduced cost and expanded generality.The method used the same sequence of dually labelled probe for all mutation sites,reducing the cost of optimization and detection.With the introduction of Sh1 and Sh2,the method could overcome complex secondary structure formed by substrates,which expanded the generality of nucleic acid probe.Combined with Endo IV,the design could reach the limit of detection of 0.05%or lower for PTENR130Q,BRCA2 rs80359065 and EGFR-L858R with synthesized single strands.Experiments on real clinical samples were conducted,and the limit of detection could be 0.1% for EGFRL858R,indicating the potential of clinical application of our design.Furthermore,we would like to point out that our design was not limited to Endo IV assisted probe,other types of nucleic acid probes could also have their cost reduced and generality improved by applying our design.In all,we believe that shared-probe system is a convenient tool for the detection of low abundance mutation and we anticipate it to be widely adopted in molecular diagnosis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos.21705053 and 81871732),the Natural Science Foundation of Hubei Province (No.2017CFB117),Hubei Province Health and Family Planning Scientific Research Project (No.J2017Q017),and Wuhan Youth Science and Technology Plan (No.2017050304010293).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.09.038.
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria