
Copper-catalyzed diversified annulations between α-diketones and alkynyl α-diketones
2022-07-11 03:39XingwenKongFngYuShengtongNiuFnGongShungYngJinggongLiuBenlongLuoXinqingFng
Chinese Chemical Letters 2022年6期
Xingwen Kong,Fng Yu,Shengtong Niu,Fn Gong,Shung Yng,Jinggong Liu,Benlong Luo,Xinqing Fng,∗
a State Key Laboratory of Structural Chemistry,and Key Laboratory of Coal to Ethylene Glycol and Its Related Technology,Center for Excellence in Molecular Synthesis,Fujian Institute of Research on the Structure of Matter,University of Chinese Academy of Sciences,Fuzhou 350100,China
b Pingxiang University,Pingxiang 337055,China
c Orthopedics Department,Guangdong Provincial Hospital of Traditional Chinese Medicine,The Second Affiliated Hospital of Guangzhou University of Chinese Medicine,Guangzhou 510120,China
Keywords:Copper catalysis α-Diketones Diversified annulations Substituents-controlled reactivities Atom economy
ABSTRACT Copper-catalyzed divergent annulations between α-diketones and alkynyl α-diketones have been achieved,delivering a series of highly functionalized and biologically important cis-hexahydro-2Hcyclopenta[b]furan (HCPF) and 2-hydroxydihydrofuran-3(2H)-one (HDFO) products with high levels of stereoselectivity under identical conditions.The protocol features the use of earth-abundant copper catalyst,mild conditions,shortening synthetic routes in constructing different molecular frameworks,and reducing the corresponding possible waste production.The substituents of the nucleophilic α-diketones play crucial roles in switching the reaction pathways.
Achieving diversified reactivity under identical reaction conditions through only slightly varying the substituents of a reactant is a charming strategy in synthetic chemistry,and accords with the criteria of green chemistry,since such a strategy significantly shortens the synthetic steps in constructing different molecular skeletons,thus reducing possible wastes and the corresponding contamination.However,such a strategy has been hard to be realized because the small change of a substituent is difficult to alter the inherent reactivity of a reaction [1–8].Compared to the more frequently used methods of getting different products through modifying reaction conditions [9–15],the aforementioned protocol is still not common and underdeveloped.
On the other hand,cis-hexahydro-2H-cyclopenta[b]furan (HCPF)and 2-hydroxydihydrofuran-3(2H)-one (HDFO) widely occur in natural products and pharmaceutically active substances (Fig.1) [16–22].Specifically,spirotrichilin A showsanti-inflammatory activity on NO production in certain cell line [16],kuhistaferone displays moderate cytotoxicity against the human tumor cell in HCT116[17],and gracilioether J has antileishmanial activity [18].Moreover,GRL-0489A displays remarkable enzyme inhibitory and antiviral potency,and its antiviral activity against multidrug resistant HIV-1 variants is even higher than FDA approved inhibitors [16];antagonists of the CC-chemokine receptor 2 (CCR2) have been vigorously pursued by many pharmaceutical companies as a target for drug discovery,because the compound could have the potential for use in the acute and chronic conditions of inflammatory and autoimmune diseases [20].However,rapid construction of both HCPF and HDFO skeletons in an efficient and stereoselective manner is still a challenge,and most of the known reports employ a sequential construction strategy to make bicyclic HCPF structures,and one-step method to form the bicycles starting from acyclic substrates remain scarce.For example,Jamison and co-workers completed the total synthesis of (-)-terpestacin,and the four-step synthesis of HCPF derivative laid the foundation of the whole process (Scheme 1a) [23,24].Similarly,two four-step methods were both used to make the bicyclic HCPF rings for the synthesis of GRL-0489A (Scheme 1b) [19]and CCR 2 antagonist (Scheme 1c)[20].In all the above case,pre-existed ring structures were used to make the second ring units and multiple steps were always needed,which rendered the synthesis of HCPF skeletons less efficient and less atom-economic.
We have been interested in disclosing the unique features ofαdiketone chemistry,and have found that under different conditions the same set of substrates can undergo distinct pathways to afford diverse products [25–27].Here we report that under the same conditions of copper catalysis the reaction ofα-diketones and alkynylα-diketones can be switched by tuning the substituent within the nucleophilicα-diketones.Copper is earth abundant,not expensive,and less toxic.Moreover,the protocol affords a series of bicyclic products containing HCPF skeletons or HDFO units,thus providing a concise approach addressing the corresponding synthetic challenges (Scheme 1d).
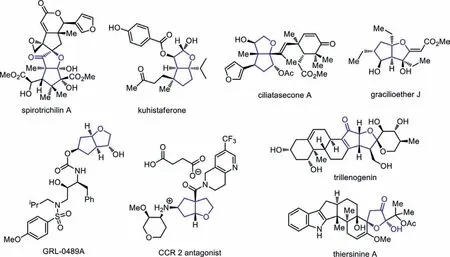
Fig.1.Natural products and bioactive compounds containing HCPF and HDFO.
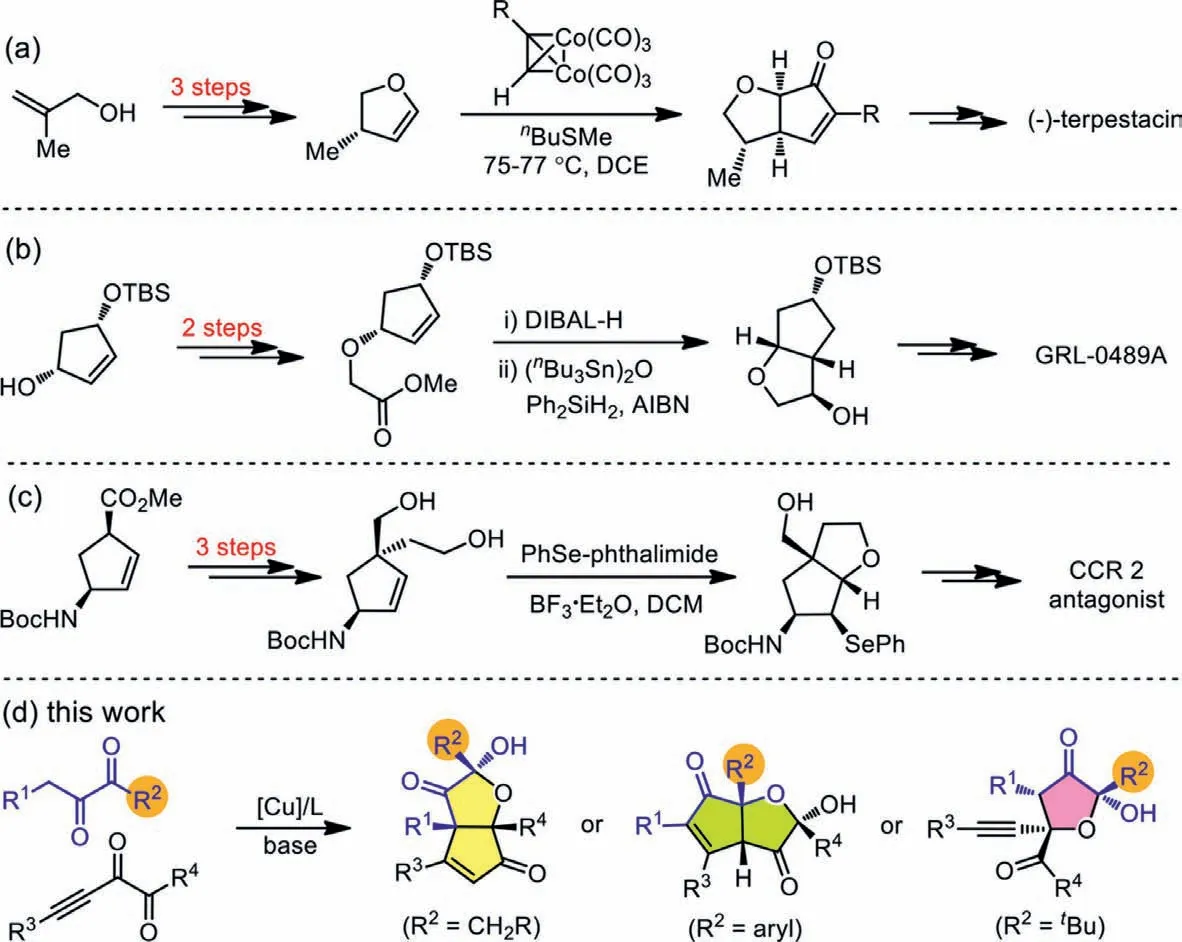
Scheme 1.Selected methods leading to HCPF compounds.
As shown in Table 1,1a and 2a were selected as the model substrates to test the corresponding annulation reactivity.Copper salts were employed to catalyze the process owing to the known abilities of copper in activating ynones [28–32].Our initial test using CuCl as the catalyst,racemicN-heterocyclic carbene L1 as the ligand,DABCO as the base,and CH2Cl2as the solvent did not afford noticeable product (Table 1,entry 1).The solvent of MeOH was also not suitable for the reaction (Table 1,entry 2).To our delight,the use of THF and toluene was able to produce 3a in 15% and 30% yield,respectively (Table 1,entries 3 and 4).3a contains acisbicyclic HCPF structure with ketone,enone,and hydroxy functionalities installed,and was obtained as the single diastereoisomer.Considering the importance of HCPF type compounds in organic synthesis and drug discovery,we continued the study to improve the yield of 3a.Using toluene as the solvent,we surveyed a series of copper catalysts such as CuI,CuCN,and CuBr2,but they could not further improve the yield of 3a (Table 1,entries 5−7).However,Cu(MeCN)4BF4and CuTc (copper(I) thiophene-2-carboxylate)showed better performance (Table 1,entries 8 and 9) and CuTc led to 3a in 65% yield.Then using CuTc,a series of other bases such as Cs2CO3,K2CO3,Et3N,DBU andiPr2NEt were tested,but no better yield of 3a was obtained (Table 1,entries 10−14).More different types of ligands such as bisphosphine L2,thiazolium L3,1,10-phenanthroline L4,and Box-ligands L5-L6 were also studied,but no higher yield was detected although most of them could deliver 3a in certain yields (Table 1,entries 15−19).Control experiments in the absence of DABCO or CuTc could not furnish 3a (Table 1,entries 20 and 21),indicating that both base and copper catalyst are necessary for the reaction.Only 24% yield of 3a was gotten without ligand (Table 1,entry 22),demonstrating that ligand is able to accelerate the annulation process.Equal amount of 1a was found to decrease the total yield of 3a (Table 1,entry 23).
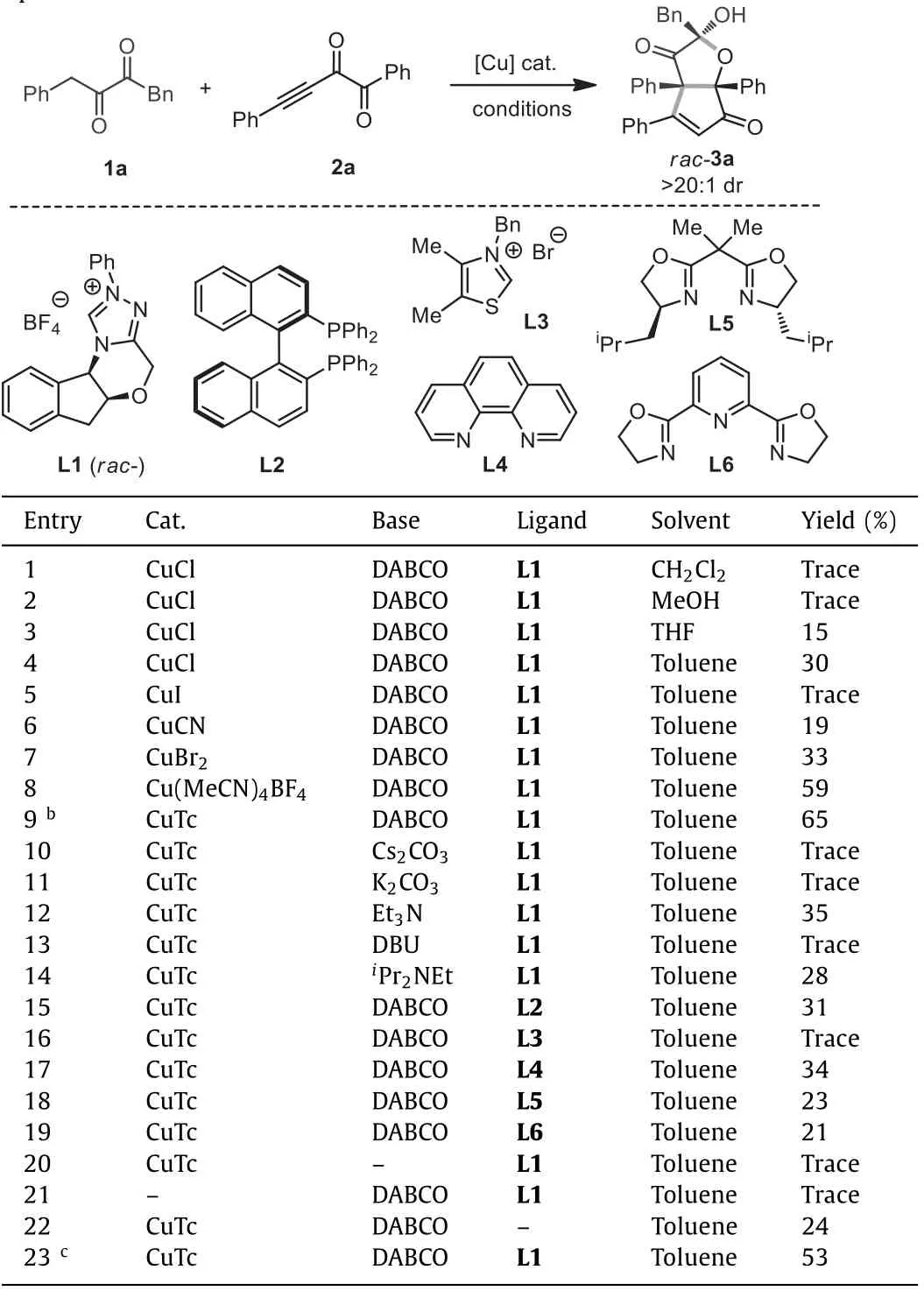
Table 1 .Optimization of reaction conditions.a
Having identified the optimal conditions,we started to examine the scope and limitation of the protocol.As shown in Scheme 2,the structure of 3a was identified clearly using X-ray single-crystal structure analysis.The variation of R3group in alkynyl diketones using electron-withdrawing or donating groups did not affect the yield,leading to the formation of 3b and 3c in good to high yields(Scheme 2,3b and 3c).Heterocyclic thienyl group showed little influence on the result (Scheme 2,3d),and aliphatic R3groups such asnhexyl and cyclopropyl proved compatible under the reaction conditions to afford 3e and 3f in 80% and 44% yields,respectively (Scheme 2,3e and 3f).Subsequently,we studied the scope of R4substituent of the alkynyl diketones.We found that when R4were phenyl groups with electron-poor F or electron-rich Me substituents the yields were not affected,delivering 3g and 3h in 62%and 68% yields,respectively (Scheme 2,3g and 3h).The reaction could also tolerate the phenyl ring bearing a carbazole substituent(Scheme 2,3i),and the use of a naphthyl group as R4proved suitable for the reaction (Scheme 2,3j).Furthermore,simultaneously varying both R3and R4groups proved possible,delivering 3k and 3l in moderate yields (Scheme 2,3k and 3l).Changing both R1and R2also proved feasible to produce 3m,albeit in relatively lower yield (Scheme 2,3m).When R1and R2groups bear different electronic properties,poor regioselectivity was observed,and both 3n and 3o were obtained with 1:1 ratio in 53% total yield (Scheme 2,3n and 3o).When R1is an aliphatic group such as Me,the corresponding diketone substrate with Bn/Et substituents (1p) has also been tested but afforded a complicated mixture of unidentified products.To be noted is that in all cases,the products were obtained as single diastereoisomers.
Further survey of the nucleophilic diketones showed that when 1 were aryl substituted diketones,a thoroughly different reaction pathway was applied for the reaction,and relatively stable bicyclic products 4a were isolated after the OH protection usingpnitrobenzoyl (PNB) group (Scheme 3,4a).The variation of R1,R2and Ar groups all proved successful,releasing 4b,4c and 4d,respectively.Both the annulation and the OH protection steps were performed in moderate yields,thus resulting the relatively low total yields.Furthermore,WhentBu-substituted diketones were studied,we found that the third mechanism predominated in the reaction,affording 2-hydroxydihydrofuran-3(2H)-one (HDFO) type products through a formal [3+2]annulation (Scheme 4,5a−5d).The reaction proved slow and certain amount of starting materials remained after 5 h.Both products 4 and 5 have three stereogenic centers and were formed with high diastereoselectivities (>20:1 dr).Moreover,4a’ and 5b were all confirmedviaX-ray singlecrystal structure analysis.Nucleophilic diketones with aliphatic R1such as Me have also been tested but showed low reactivities in forming the corresponding annulation products.Alkynyl diketones with alkyl R3substituent such as Me failed to deliver the corresponding products,and side reactions were detected.
A series of further transformations using 3a can be achieved.As shown in Scheme 5,an unexpected rearrangement occurred when 3a was treated by PhMgBr,and after OH protection,6a was formed with high diastereoselectivity.In this reaction,the Grignard reagent might act as a strong base to facilitate the ring opening of 3a,and the corresponding intermediate then undergoes a similar transformation as that occurs in the formation of 4.The protection of 3a afforded 6b in 69% yield and dibromide 6c could be produced from 6b in good yield with the treatment of Br2.Debromination of 6c under transition-metal catalysis allowed access to 6d The configuration of 6a,6c and 6d were all confirmed unambiguouslyviaX-ray structure analysis.
The possible mechanisms for the diversified formation of three types of products are shown in Scheme 6.For product 3a,the deprotonation of 1a leads to enolate I-1,and after addition of I-1 to 2a,allenolate I-2 is formed.A new type of enolate I-3 is produced from the proton transfer within I-2,and the following aldol reaction initiates a formal [3+2]annulation to release I-4,and after protonation,3a is obtained (Scheme 6a).As to the formation of 4a(Scheme 6b),after the Michael addition of enolate II-1 to 2a,II-2 is formed,and the attack of phenyl ketone by the allenolate gives II-3.Then after the intramolecular annulation and protonation,II-5 is generated.The isomerization happens for II-5 to deliver more stable product 4a.tBu-substituted 1g also produces enolate species III-1 as the first step,but then the internal carbonyl of 2a is attacked by III-1,and after a formal [3+2]annulation and protonation,5a is formed (Scheme 6c).Because the reaction cannot occur without CuTc catalyst,we postulate that copper salt is to activate the alkynyl diketones [32–34].As to the mechanism divergence between 1a and 1d,after the initial Michael additions,phenyl ketone seems to be more active than benzyl ketone,thus is attacked by the allenolate intermediate.
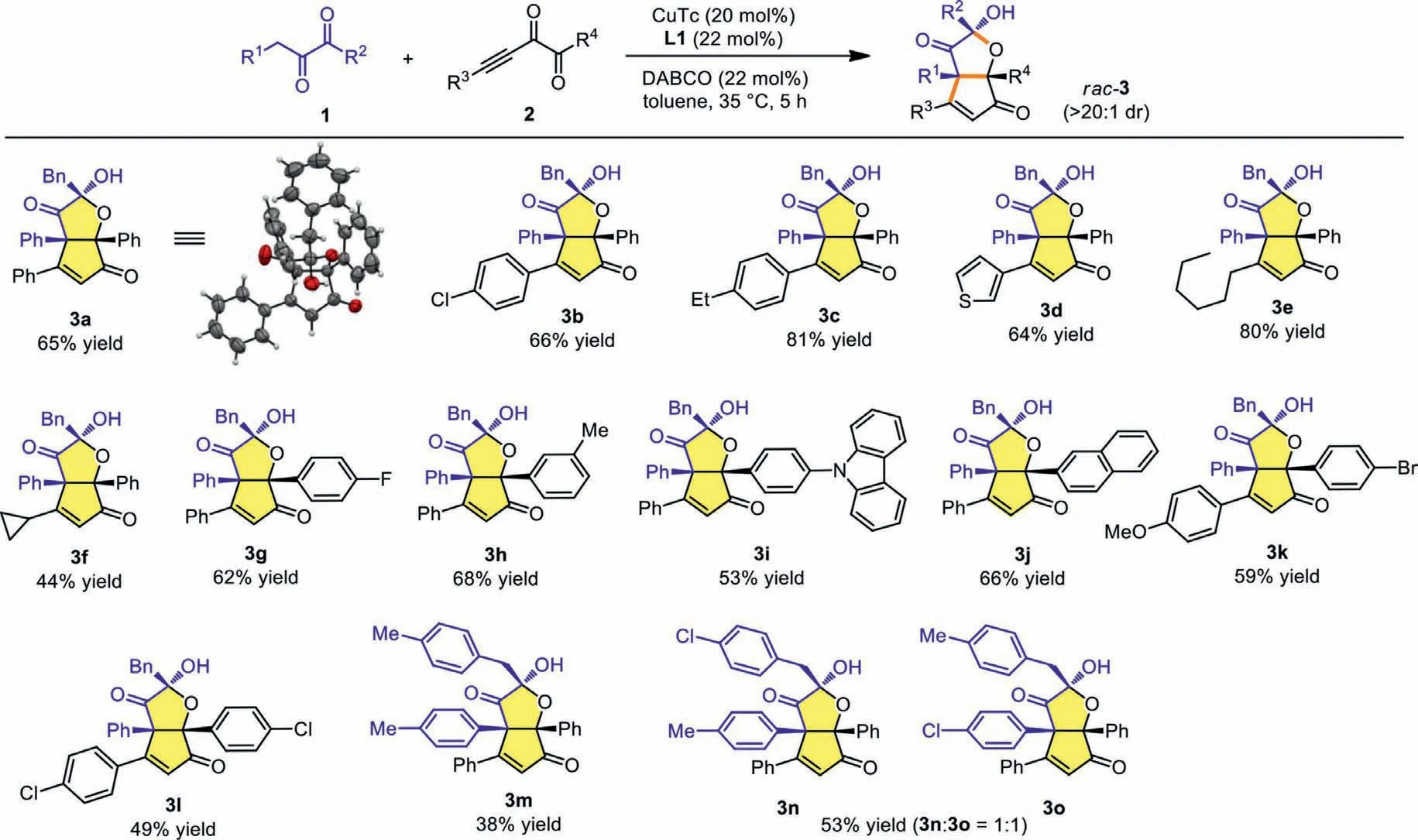
Scheme 2.Scope of functionalized HCPF 3 formation.Reactions conditions: 1 (0.3 mmol),2 (0.2 mmol),CuTc (20 mol%),DABCO (22 mol%),L1 (22 mol%),toluene (1 mL),35 °C,5 h,under argon atmosphere.The diastereomeric ratios were determined via 1H NMR analysis of the reaction mixtures.All isolated yields were based on 2.
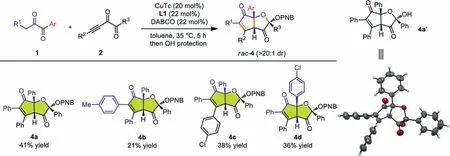
Scheme 3.Scope of functionalized HCPF 4 formation.Reactions conditions for annulations: 1 (0.3 mmol),2 (0.2 mmol),CuTc (20 mol%),DABCO (22 mol%),L1 (22 mol%),toluene (1 mL),35 °C,5 h,under argon atmosphere.Reaction conditions for OH protection and HCPF 4 formation: p-nitrobenzoyl chloride (PNB-Cl,0.4 mmol),DMAP (20 mol%),DABCO (2 equiv),CH2Cl2 (1 mL),r.t.,5 h,under argon atmosphere.The diastereomeric ratios were determined via 1H NMR analysis of the reaction mixtures.All yields were isolated yields based on 2.
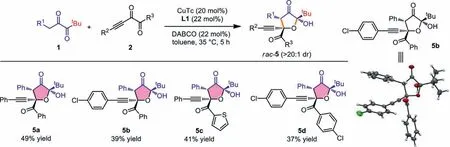
Scheme 4.Scope of functionalized HDFO 5 formation.Reactions conditions: 1 (0.3 mmol),2 (0.2 mmol),CuTc (20 mol%),DABCO (22 mol%),L1 (22 mol%),toluene (1 mL),35 °C,5 h,under argon atmosphere.The diastereomeric ratios were determined via 1H NMR analysis of the reaction mixtures.All yields were isolated yields based on 2.
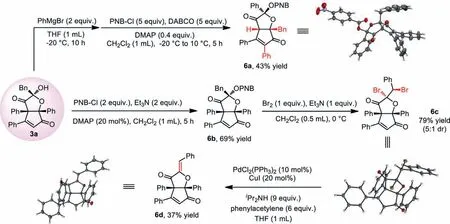
Scheme 5.Synthetic applications of 3a.
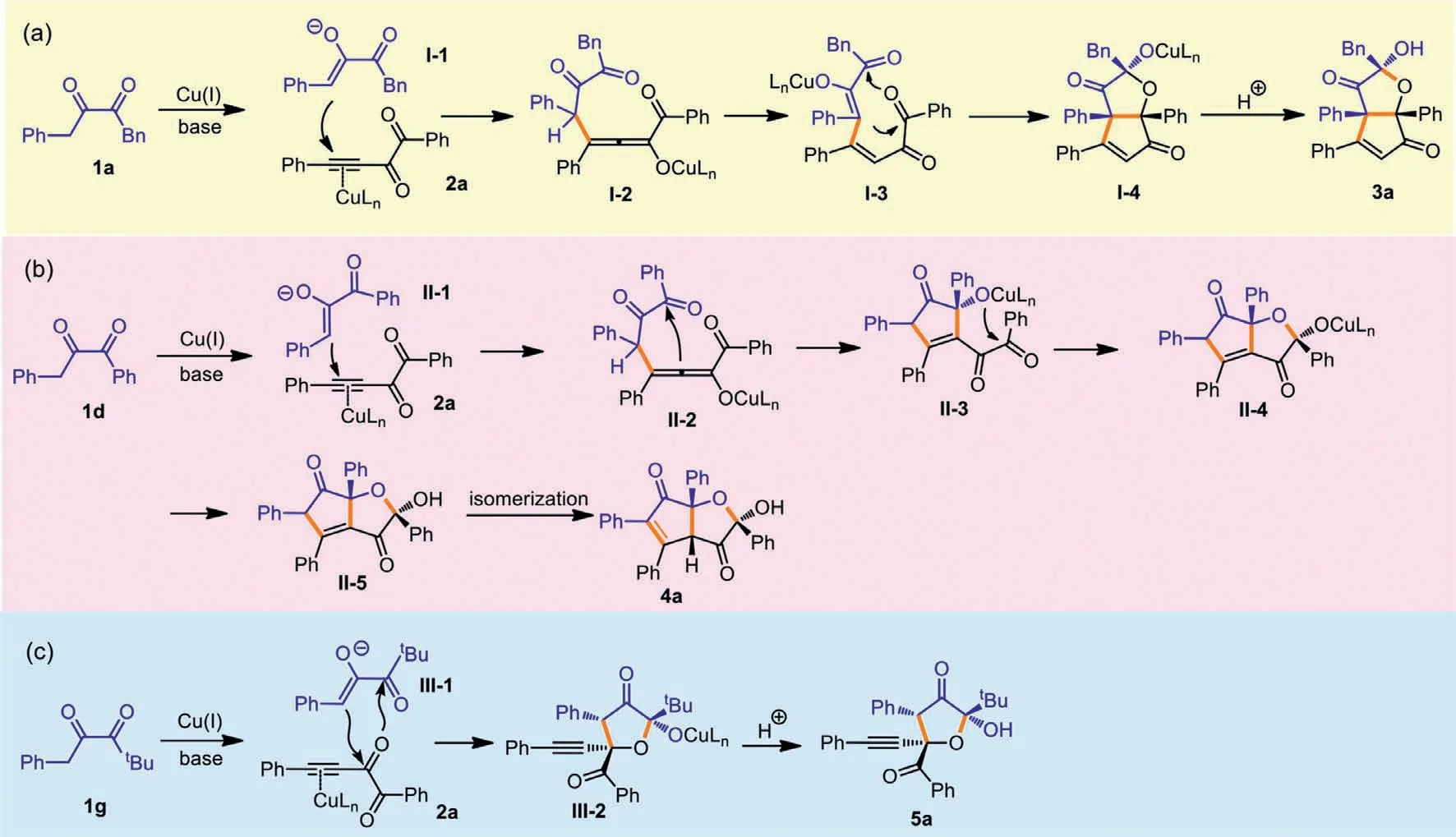
Scheme 6.Postulated mechanisms.
In summary,copper-catalyzed divergent annulations betweenα-diketones and alkynylα-diketones have been developed,and a series of highly functionalizedcis-hexahydro-2Hcyclopenta[b]furan (HCPF) and 2-hydroxydihydrofuran-3(2H)-one(HDFO) products are obtained with high levels of stereoselectivity control under mild conditions.The substituents of the nucleophilic α-diketones play crucial roles in determining the reaction pathways.All three types of products were obtained in an atom-economic fashion,and the protocol greatly reduces the synthetic steps and the corresponding possible waste production.Further studies of developing an enantioselective version of this work and revealing the new chemistry of α-diketone compounds are ongoing in our group.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos.22071242 and 21871260),the Strategic Priority Research Program of the Chinese Academy of Sciences (No.XDB20000000),Fujian Natural Science Foundation (No.2018J05035),and Science and Technology Research Program of the Education Department of Jiangxi Province (No.GJJ1991151).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.11.070.
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria