
Rhodium-catalyzed formal [4+3]annulation reaction of N-methoxybenzamides with gem–difluorocyclopropenes: A combination of experimental and theoretical studies
2022-07-11 03:39YimioHeLimeiTinXuexueChngZemingQuYnminHungChushengHungQingSunHonggenWng
Chinese Chemical Letters 2022年6期
Yimio He,Limei Tin,Xuexue Chng,Zeming Qu,Ynmin Hung,Chusheng Hung,Qing Sun,Honggen Wng
a Guangxi Key Laboratory of Natural Polymer Chemistry and Physics,College of Chemistry and Materials,Nanning Normal University,Nanning 530001,China
b Key Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle,Nanchang Hangkong University,Nanchang 330063,China
c School of Pharmaceutical Sciences,Sun Yat-sen University,Guangzhou 510006,China
Keywords:gem-Difluorocyclopropene Fluorinated 2H-azepin-2-one C-H functionalization Oxidizing directing group Rhodium[4+3]Annulation
ABSTRACT A rhodium-catalyzed [4+3]cycloaddition reaction between N-methoxybenzamides and gem–difluorocyclopropenes is described.The reaction offers a mild and efficient approach towards the synthesis of fluorinated 2H-azepin-2-ones with broad substrate scope.A consecutive HOAc-assisted C–N bond formation and fluorine elimination are involved as key steps for success as illustrated by detailed DFT studies.
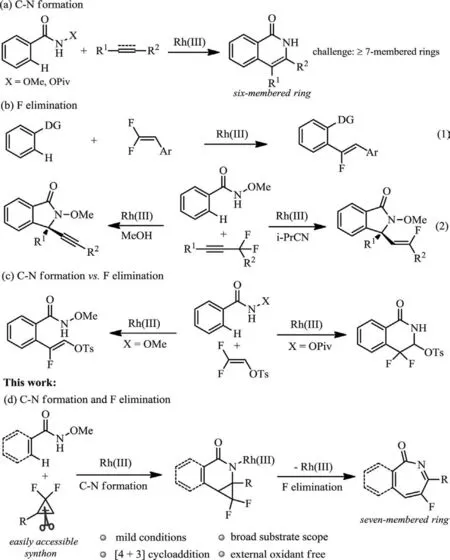
Transition metal-catalyzed C–H bond functionalization reactions are among the most straightforward and atom-economic synthetic methodologies for the construction of complex molecules [1–4].Typically,a directing group is need to facilitate a regioselective C–H activation event.And the use of oxidizing directing group has received tremendous attentions by offering enhanced reactivity and eliminating the employment of external oxidant [5–12].In this respect,various five-and six-numbered rings are mildly and effectively constructed through the oxidizing directing group strategy.For example,an elegant seminal work from Fagnou reported a Rh(III)-catalyzed redox-neutral annulation of benzhydroxamic acids with alkynes towards the synthesis of isoquinolone derivatives by using N–O bond as a built-in oxidant (Scheme 1a) [13].However,since the eight-membered rhodacycle intermediates are energetically unstable,utilizing this tactics to construct seven-membered rings is elusive [14,15].
Fluorinated organic molecules have attracted significant attention in drug discovery and agricultural chemistry due to their unique physicochemical and bioactivity properties [16–18].Traditional methods for the incorporation of fluorine into the molecules often suffer from the need of substrate pre-activation,the use of non-readily available starting materials,low regio-or stereoselectivity and/or poor functional group tolerance due to the employment of sensitive reagents [19–21].Compared with the abovementioned protocols,transition metal-catalyzed C–H/C-F bond activation assisted by a directing group provides a concise and reliable alternative in an atom-and step-economic pattern.In this context,the group of Loh reported a RhIII-catalysed tandem C–H/C-F activation for the synthesis of (hetero)arylated monofluoroalkenes usinggem–difluoroalkenes as electrophiles (Scheme 1b-1) [22–24].The group of Wang disclosed a solvent-dependent enantioselective synthesis of alkynyl and monofluoroalkenyl isoindolinones by asymmetric CpRhIII-catalyzed C–H activation withα,α-difluoromethylene alkyne as the substrate (Scheme 1b-2) [25–28].In these two cases,metal-mediatedβ-fluorine elimination was observed as key step.Previously,we discovered that different directing groups (N–OMe andN–OPiv amides) enabled dictate the selectivity of C–N formationversus β-F elimination with 2,2-difluorovinyl tosylate as a substrate (Scheme 1c) [29–33].WithN–OMe benzamide being a directing group (DG),the reaction delivered a monofluorinated alkene with the retention of the tosylate functionality.WhenN–OPiv benzamides were used,however,[4+2]cyclization occurred to providegem–difluorinated dihydroisoquinolin-1(2H)-ones.
Scheme 1.Rhodium-catalyzed C–H activation reactions toward the organofluorines with an oxidizing directing group.
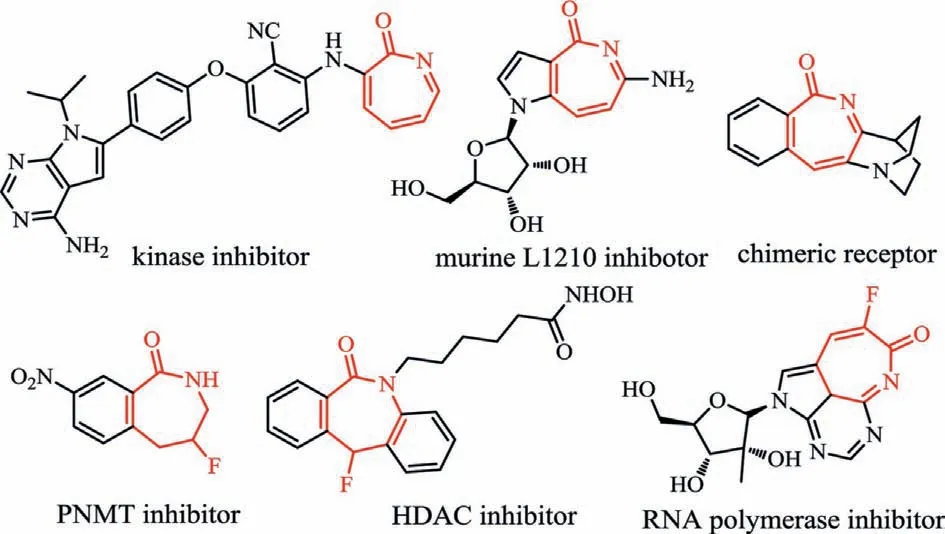
Fig.1.Representative bioactive molecules.
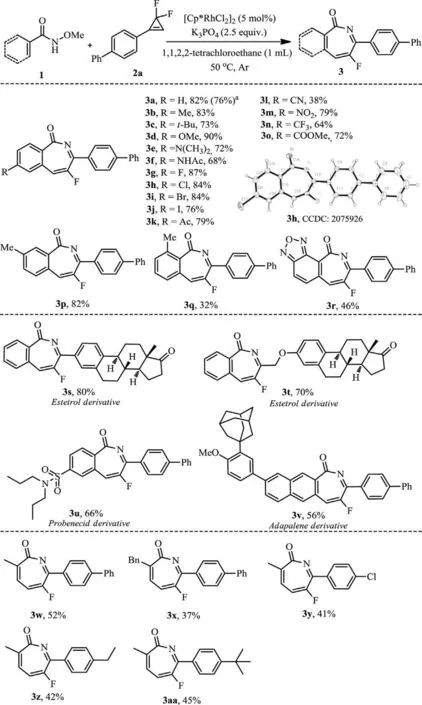
Scheme 2.Substrate scope on amides.Reaction conditions: 1 (0.2 mmol),2(0.24 mmol),[Cp∗RhCl2]2 (5 mol%),K3PO4 (2.5 equiv.),1,1,2,2-tetrachloroethane(1 mL) at 50 °C under argon atmosphere for 12 h.Isolated yield.a 1a (10 mmol),2a (12 mmol),[Cp∗RhCl2]2 (5 mol%),K3PO4 (2.5 equiv.),1,1,2,2-tetrachloroethane(10 mL) at 50 °C under argon atmosphere for 12 h.
Herein,we report a rhodium-catalyzed formal [4+3]cycloaddition reaction ofN-methoxybenzamides with easily accessiblegem–difluorocyclopropenes [34–49].The reaction allows the formation of highly functionalized fluorinated 2H-azepin-2-one frameworks with excellent regioselectivity and functional group tolerance (Scheme 1d).Some interesting features of the transformation include: i) Both C–N bond formation and fluorine elimination occur in the reaction withN–OMe as an internal oxidant;ii) The combination of [4+2]cycloaddition and retro-[2+1]strategy eliminates the formation of eight-membered rhodacycle,thereby providing a feasible and reliable route for the construction of sevenmembered aromatic heterocycles;iii) This reaction proceeds under rather mild conditions,and a series of bioactive fluorinated 2Hazepin-2-one derivatives (Fig.1) [50–52]are obtained in moderate to good yields.
The reaction was initially investigated by usingNmethoxybenzamide 1a andgem–difluorocyclopropene 2a as model substrates,[Cp∗RhCl2]2as catalyst and K3PO4as base in CH2Cl2at 50 °C under argon atmosphere.To our delight,the desired product 3a was obtained in 41% yield (Table 1,entry 1).Solvent screening revealed that only chlorinated alkanes promoted the transformation,with 1,1,2,2-tetrachloroethane being the solvent of choice to give a high yield of to 82% (Table 1,entries 2–7).Replacing [Cp∗RhCl2]2with Cp∗Rh(OAc)2led to a reduced yield of 61% (Table 1,entry 8).The reaction did not proceed with other transition metal catalysts such as [Cp∗IrCl2]2,[RuCl2(p-cymene)]2and [CoCp∗(CO)I2](Table 1,entries 9–11).Other inorganic bases were also subsequently used in the reaction,however,the yield of 3a was not further improved (Table 1,entries 12–16).
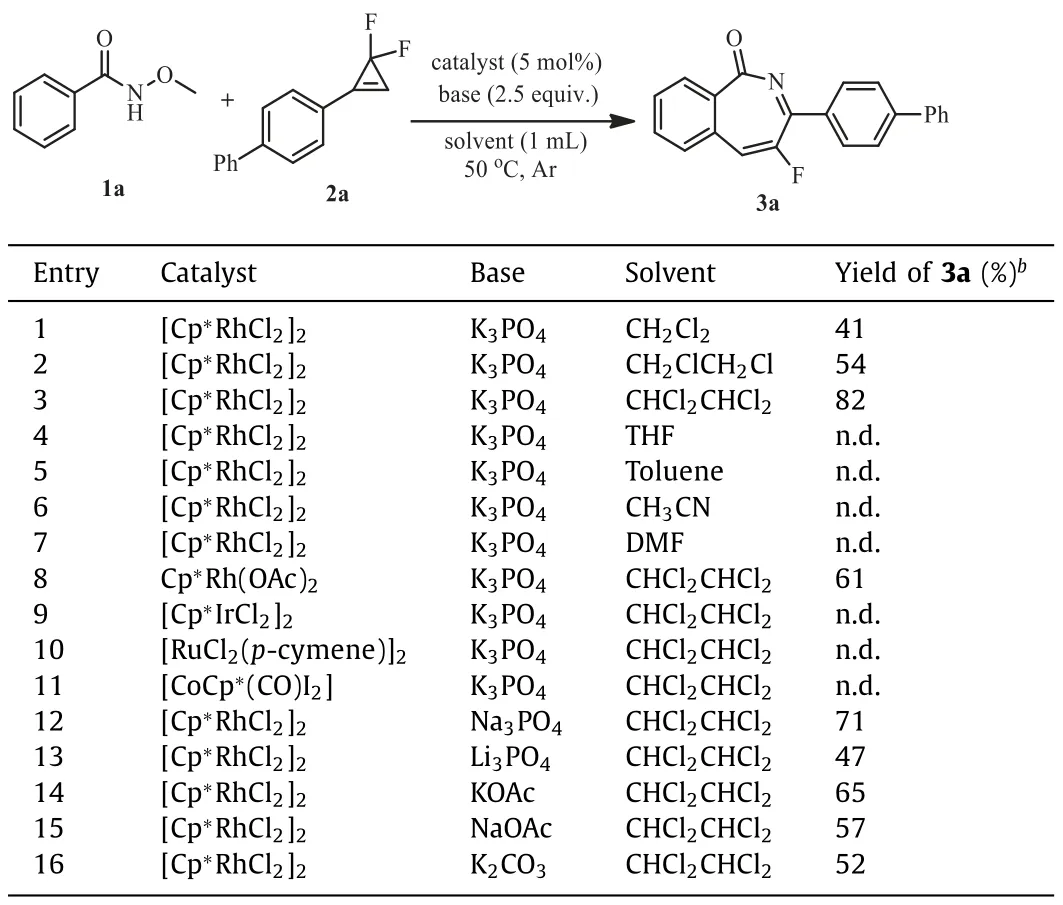
Table 1 Optimization of reaction conditions.a
With optimized conditions in hand,we set to examine the scope.As shown in Scheme 2,the aromatic amide substrates bearing electron-donating (such as Me,t-Bu,OMe and N(CH3)2)and electron-withdrawing substituents (such as Ac,CN,NO2,CF3,CO2Me) atparaposition underwent reaction smoothly,delivering the cyclized products 3a-3f and 3k-3o in good yields.Substrates bearing halogen substituents were also compatible well(3g-3j),thus providing valuable handles for follow-up transformations.Whenmeta-substitutedN-methoxyamide 1p was used,the rhodation occurred at the less hindered site to provide exclusively the C-6 substituted regioisomer (3p).Specially,the 2-methyl substituted substrate did not retard the process (3q),although the yield was slightly reduced.To further highlight the synthetic versatility of our method,several substrates derived from complex natural products and drugs were also subjected to the reaction and the corresponding fluorination products were obtained without difficulty (3s-3v).Furthermore,various alkenyl amides with 2-alkyl substitution were also feasible substrates,producing the products 3w-3aa in moderate yields.Out of our expectation,N–methoxy-2-phenylacrylamide showed no reactivity in the protocol.To demonstrate the scalability of this methodology,the reaction of 1a with 2a was performed on 10 mmol scale,affording 76% yield of the product 3a.
The substrate scope forgem–difluorocyclopropenes was also explored (Scheme 3).It was found the reaction was not sensitive to the electron nature of the substituents on the aryl ring,as a diverse of substituents such as Me,n-Pr,F,Cl,Br,CF3,NO2and CO2Me well survived in the reaction (3ab-3al).Interestingly,higher yields were obtained formeta-substituted arylgem–difluorocyclopropenes (3ai-3al).The observed higher bench stability ofmeta-substituted arylgem–difluorocyclopropenes could be a reason for this result.Not unexpectedly,substrates with other aromatic heterocycles,for example,thiochroman,pyridine,thiophene and benzoxazole were also tolerant,obtaining the products 3an-3aq in 44%−65% yields.It was intriguing that alkyl-substitutedgem–difluorocyclopropenes were also compatible (3ar-3av),greatly expanding the diversity of the title reaction.
Also interesting was the applicability ofNmethoxybenzothioamide 4 in the reaction.The cyclization reaction proceeded smoothly to give the desired product 5 in 73% yield(Scheme 4).
WhenN–methoxy-2-naphthamide 6 was used in the reaction,the 3-position C–H bond with less steric hindrance was exclusively functionalized to give the product 7 in 72% yield (Scheme 5a).Interestingly,treatment ofN-methoxybenzo[d][1,3]dioxole-5-carboxamide 8 with 2a provided the 4-position annulation product 9 in 78% yield (Scheme 5b).The later could be explained by a coordination effect between the oxygen atom at the 3-position and the transition metal.
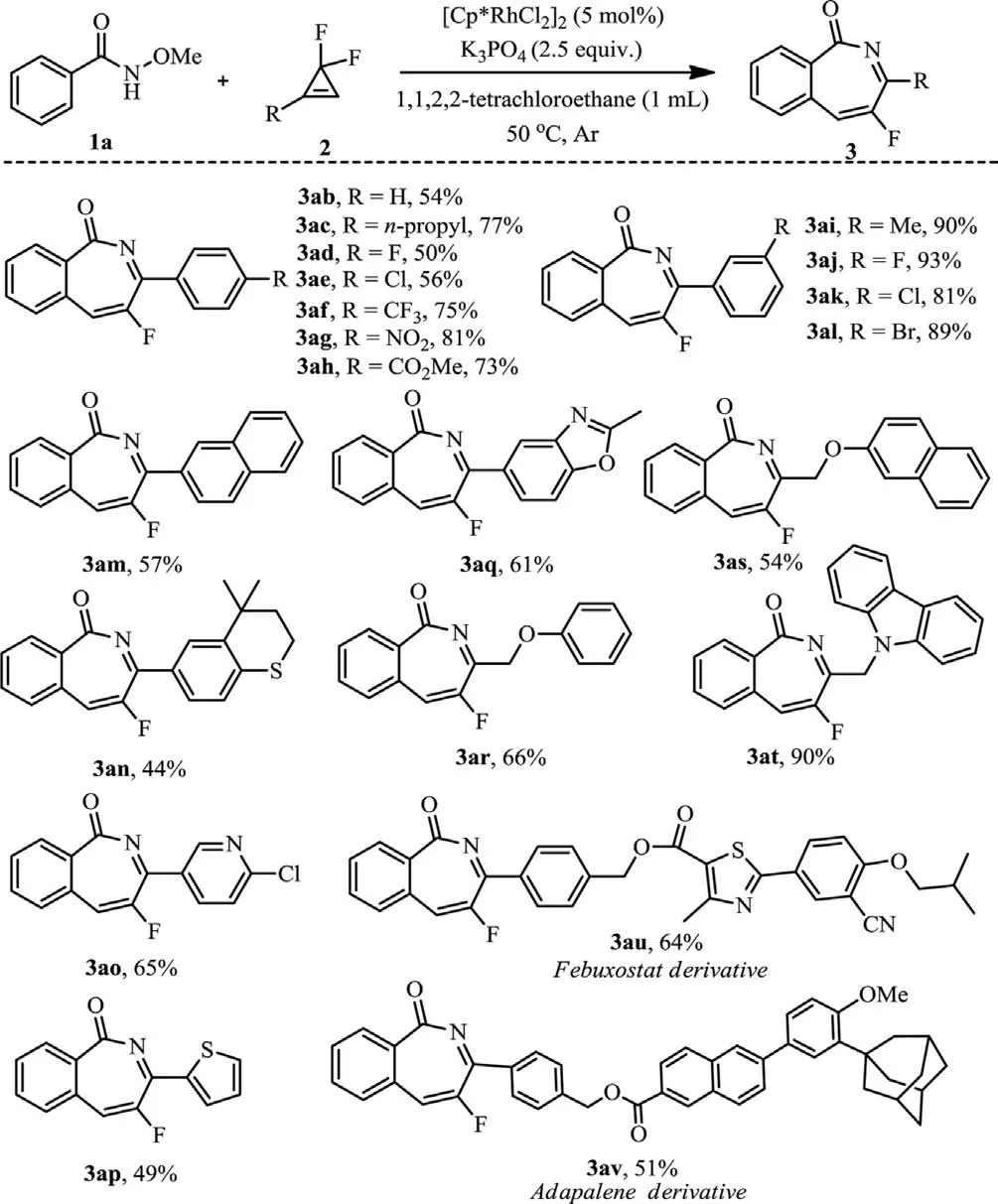
Scheme 3.gem–Difluorocyclopropene scope.Reaction conditions: 1 (0.2 mmol),2 (0.24 mmol),[Cp∗RhCl2]2 (5 mol%),K3PO4 (2.5 equiv.),1,1,2,2-tetrachloroethane(1 mL) at 50 °C under argon atmosphere for 12 h.Isolated yield.
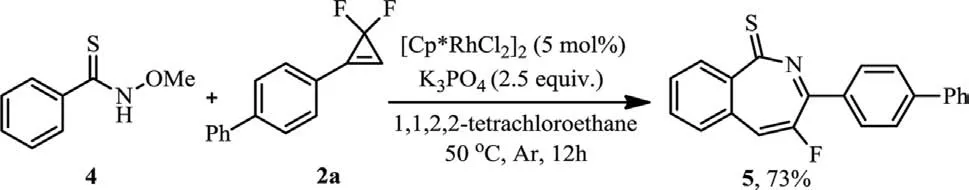
Scheme 4.Annulation reaction of 4 with 2a.
Intermolecular competitive reactions were performed to understand the reactivity ofN-methoxybenzamides andgem–difluorocyclopropenes (Scheme 6).Treatment ofNmethoxybenzamide 1a andN-methoxybenzothioamide 4 with 2a under the standard conditions gave only 3a in 80% yield (Scheme 6a).Competition reaction of 4–methoxy-N-methoxybenzamide 1d and 4-acetyl-N-methoxybenzamide 1k with 2a gave exclusively 3k in 76% yield (Scheme 6b).This result demonstrated that the benzamide substrates with electron-donating substituents are less reactive.Furthermore,when 1a was treated with 2ac and 2ah,the corresponding products 3ac and 3ah were isolated in 13% and 68%yields,respectively,suggesting that electron-poorer 2ah is good for the reaction (Scheme 6c).
To further probe the mechanism,several control experiments were conducted (Scheme 7).When D2O was added to the reaction in the absence of 2a,a 47% deuterium incorporation atorthoposition of 1a was observed without N–O bond cleavage (Scheme 7a).And a kinetic isotope effect (KIE) value ofkH/kD=1.3 was observed(Scheme 7b).These results suggested that the C–H bond cleavage is reversible and not be involved in the turnover-limiting step.A hydroamination product 10 was unexpectedly obtained in the absence of rhodium catalyst (Scheme 7c).However,this compound was demonstrated not to be an effective intermediate for the title reaction.A rhodacycle Rh-1 was prepared and its intermediacy in the reaction was confirmed by stoichiometric and catalytic reactions,suggesting a C–H activation took place.
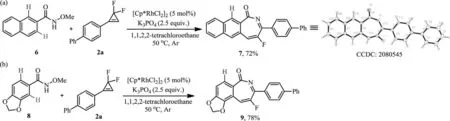
Scheme 5.Regioselective reactions.
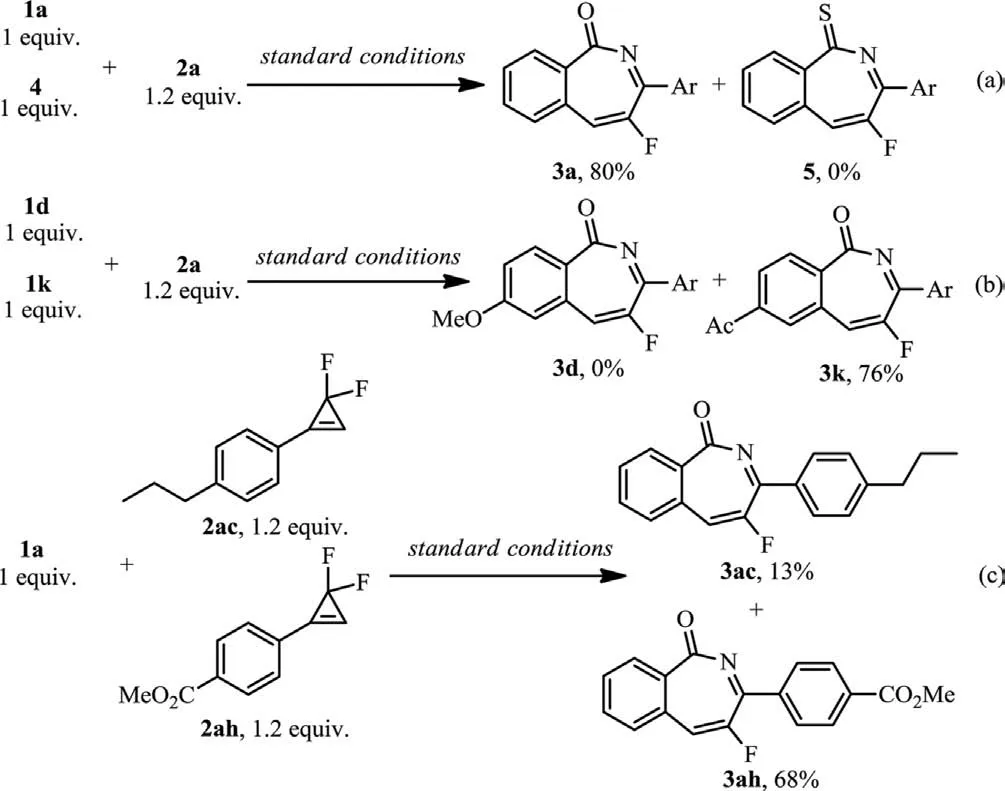
Scheme 6.Intermolecular competitive reactions.
To further cast light on the mechanism,theoretical calculations were performed at the density functional theory level(B3LYP).For the convenience of calculation,the active catalyst Cp∗Rh(OAc)2was chosen as the starting point (zero value of energy).UsingN–methoxy benzamide 1a as a substrate,N–H deprotonation followed by C–H activation were performedviaa concerted metalation-deprotonation (CMD) mechanism with acetate acting as intramolecular base,through transition states TS-1(ΔG‡=13.0 kcal/mol) and TS-2 (ΔG‡=19.8 kcal/mol),respectively(Fig.2).The intermediate verification experiments in Scheme 7c echoed the calculation results.Thereafter,the insertion ofgem–difluorocyclopropene 2ai into the rhodacycle INT-5 presented two characteristic spatial arrangements,TS-3 (ΔG‡=27.5 kcal/mol) and TS-3′(ΔG‡=28.9 kcal/mol),both of which had a higher activation barrier than the first two steps (Fig.3).The computational results indicated that C–H activation was not the turnover limiting step in the reaction,consistent with the observed small experimental KIE values (Scheme 7b).Taking into account the higher energy barrier of TS-3′,especially TS-4′,therefore subsequent calculations revolved around TS-3.From INT-7,the priority of eitherβ-fluorine elimination or C–N bond formation was discussed.The results revealed that the directβ-fluorine elimination with or without the assistance of acetic acid,followed by C–N bond formation stepviaTS-4a and TS-4b,featured a high energy barrier of 33.2 and 35.1 kcal/mol,reaspectively.Two possible pathways for C–N bond formation prior to the defluorination were then calculated.Considering the high energy barrier of TS-4c (ΔG‡=68.6 kcal/mol),the direct migration of the methoxy group from the amide to the trivalent rhodium to form INT-9c was tough.The migration process was more reasonable in the assistance of acetic acid,because the energy barrier of TS-4 was reduced to 28.8 kcal/mol and a Rh(V) intermediate INT-10 was produced with the free-energy of −54.3 kcal/mol.the synergistic effect of rhodium and acetate accelerated the ring-opening defluorination of INT-10 to release the final product 3ai (ΔG‡=−71.0 kcal/mol).Overall,the computed Gibbs free-energy changes of the reaction pathway demonstrated a redox-neutral Rh(III)-Rh(V)-Rh(III) catalytic cycle for the developed protocol involving HOAc-prompted oxidative addition and unprecedented C-F bond cleavage/ring expansion processes.
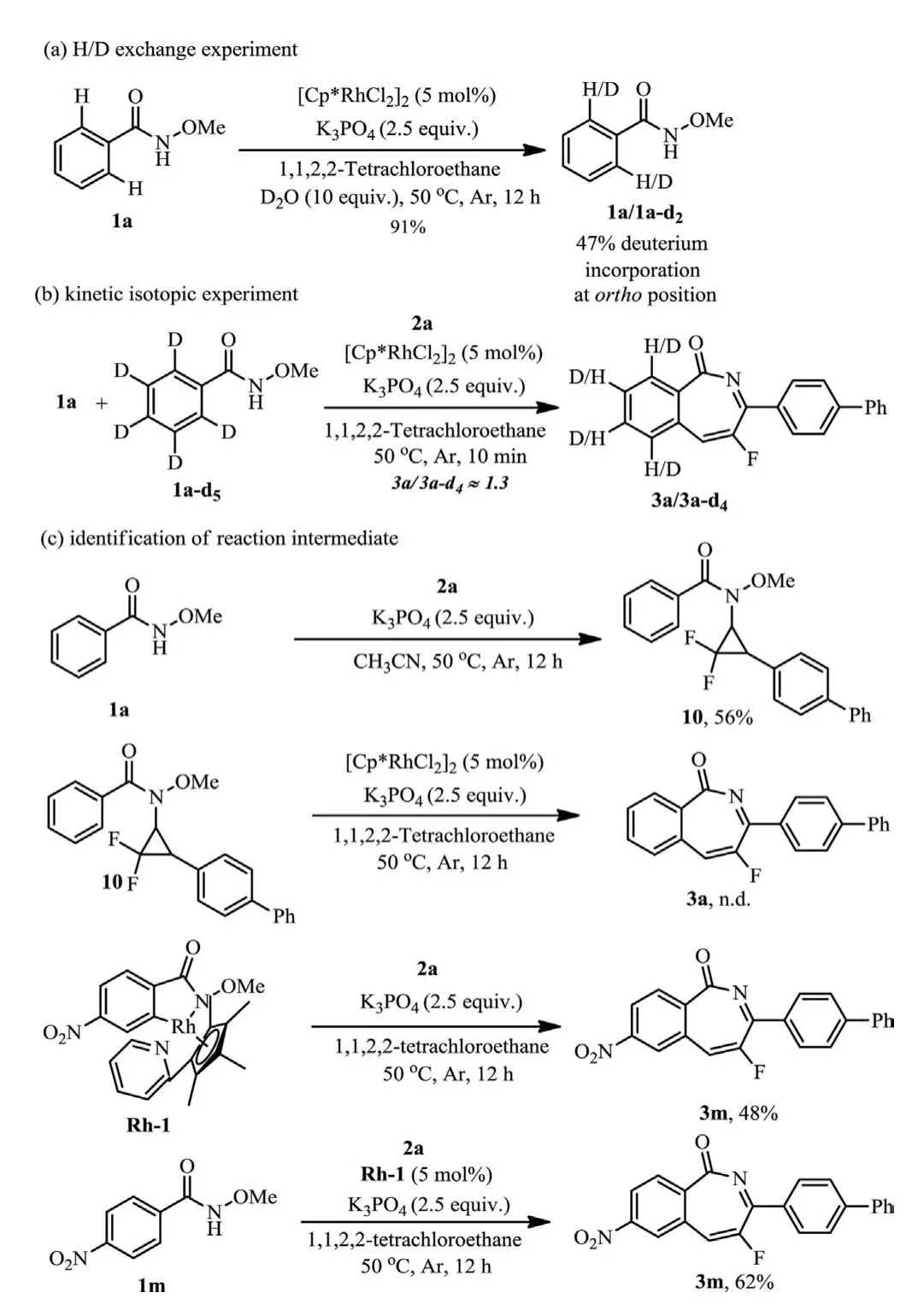
Scheme 7.Mechanism studies.
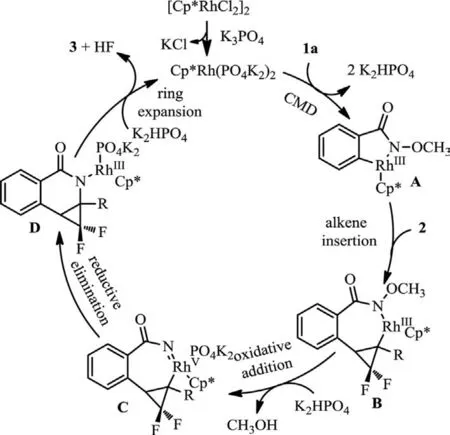
Scheme 8.Possible catalytic cycle.
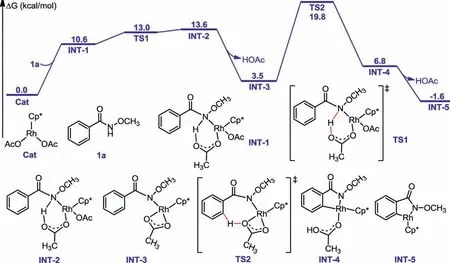
Fig.2.Computed pathways for N–H and C–H activation.
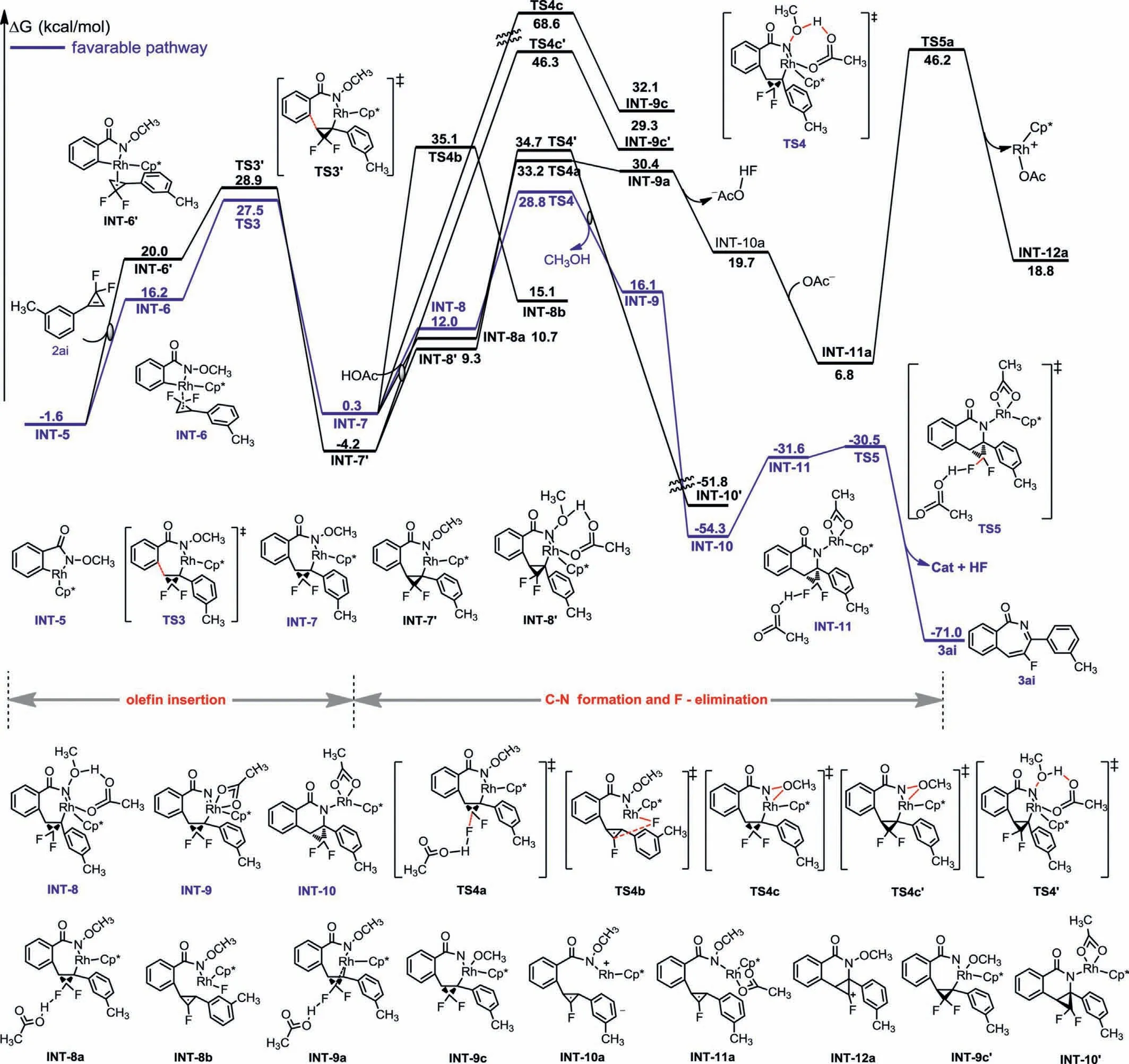
Fig.3.Computed pathways for C–N formation and F-elimination.
On the basis of the above studies and previous reports [53–57],a plausible mechanism is proposed in Scheme 8.A ligand exchange between [Cp∗RhCl2]2and K3PO4forms a reactive catalyst.Rhodacycle A is then formedviaconsecutive N–H and ortho C–H bonds activation.These processes may occurviaa CMD (concerted metalation deprotonation)-like mechanism in the aid of internal PO43−base [29].Afterwards,a migratory insertion of the rhodacycle A intogem–difluorocyclopropene 2 delivers the intermediate B.Rh(III) in intermediate B is oxidized to Rh(V) nitrenoid intermediate C in the aid of K2HPO4.Rh(V) intermediate C returns to Rh(III) through a C–N migratory insertion into the nitrenoid.Finally,the synergistic effect of rhodium and K2HPO4accelerates the ring-opening defluorination to release the product 3.
In summary,we developed a novel [4+3]cycloaddition reaction ofN-methoxyamides withgem–difluorocyclopropenes,enabling a modular,concise and efficient approach for accessing highly functionalized fluorine-substituted 2H-azepin-2-ones in moderate to good yields.Other appealing features include simple and readily available substrates,mild conditions and broad substrate scope.DFT studies revealed a consecutive C–N bond formation and fluorine elimination events in the annulation reaction.Given the importance of 7-membered heterocycles as well as fluorine atom in medicinal chemistry,we anticipate this protocol will find applications.During the preparation of this work,Yi and Zhou reported a similar work [58].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (Nos.21861007,21702034),Natural Science Foundation of Guangxi Province (No.2021GXNSFAA075024),“BAGUI Scholar” Program of Guangxi Province of China,High-Level Innovation Team and Distinguished Scholar Program in Guangxi Colleges and Universities.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.01.068.
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria