
Recent advances of two-dimensional CoFe layered-double-hydroxides for electrocatalytic water oxidation
2022-07-11 03:38YiZhouJiliHuLichunYngQingshengGo
Chinese Chemical Letters 2022年6期
Yi Zhou,Jili Hu,Lichun Yng,Qingsheng Go,∗
a College of Chemistry and Materials Science,and Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications,Jinan University,Guangzhou 510632,China
b School of Materials Science and Engineering,and Guangdong Provincial Key Laboratory of Advanced Energy Storage Materials,South China University of Technology,Guangzhou 510640,China
c Ningbo Fengcheng Advanced Energy Materials Research Institute Co.,Ltd.,Ningbo 315000,China
Keywords:CoFe layered-double-hydroxides Electrocatalytic water oxidation Two-dimensional nanostructures Surface reconstruction d-band
ABSTRACT Oxygen evolution reaction (OER) is pivotal to drive green hydrogen generation from water electrolysis,but yet is strictly overshadowed by the sluggish reaction kinetics.Earth-abundant and cut-price transitionmetal compounds,particularly CoFe layered-double-hydroxides (LDHs),show the distinct superiorities in contrast to noble metals and their derivatives.In this review,we firstly underline their fundamental issues in electrocatalytic water oxidation,including CoFe LDHs crystal structure,the surface of (hydr)oxides confined to OER and the controversial roles of Fe species,aiming at understanding the structure-related activity and catalytic mechanism.Advanced approaches for optimizing OER activity of CoFe LDHs are then comprehensively overviewed,which will shed light on the different working mechanisms and provide a concise analysis of their unique advantages.Finally,a perspective on the future development of CoFe LDHs electrocatalysts is offered.We hope this review can give a concise and explicit guidance for the development of transition-metal-based electrocatalysts in the energy field.
1.Introduction
The demand of sustainable energy technologies is drastically growing due to unregulated environmental contamination and superfluous CO2emission from the over consumption of fossil fuel[1].Water electrolysis,coupling oxygen evolution reaction (OER) at anodes with hydrogen evolution reaction (HER) at cathodes,finds a promising way out of the current dilemma [2–6].However,the 4-electron uphill reaction pathway of OER unfortunately leads to a substandard efficiency of overall water splitting.Accordingly,anodic OER is treated as a pivotal entry point for uncovering the knack of commercial water electrolysis [7–10].
Nowadays,chemists and materials scientists devote extensive efforts in exploiting transition-metal compounds with superior OER performance,for the replacement of noble metals and their derivatives that are severely prohibited by the scarcity and high cost [11–13].Among first-row 3d transition-mental (Fe,Co and Ni) compounds,Co and its derivatives have attracted particular research interest owing to the prominent intrinsic OER activity and long-term durability in a large pH range (neutral to strong alkaline),in contrast to Ni based electrocatalysts (pH 7-11)whose surface deprotonation is obviously impacted by the electrolyte pH [14–16].Apart from aforesaid merits,hydrotalcite-like CoFe layered-double-hydroxides (LDHs) are generally electrolytepermeable,affording a large amount of active sites toward efficient OER.Besides,the layered structure offers an approach for preparing few-layered CoFe LDHsviaexfoliation of the raw materials,which gives rise to an ample amount of active sites,convertible electron configuration,and more distorted and defective structures[17–19].For instance,Wang and co-workers fabricated highly active monolayer CoFe LDHs with multiple vacanciesviaetching bulk CoFe LDHs through Ar plasma [20].Moreover,the synergistic effects between Fe and Co sites are favored in OER,and Hunget al.identified that Fe stabilizes Co ions under a higher valence state during OER and promotes the generation of more stable reactant intermediates [21].Besides,Fe species have highest intrinsic activity and might severs as dynamical active sites over the CoOxHyhost [22].Therefore,CoFe LDHs possess a high intrinsically catalytic activity,and outperform other Co based LDHs (e.g.,CoNi and CoMn,LDHs) which are unintentionally contaminated by Fe impurities due to using unpurified basic electrolytes,leading to complicated catalytic mechanism.In addition,Dionigiet al.and Gaoet al.,have an excellent summary about NiFe-Based catalysts for(photo)electrochemical water oxidation [23,24],while CoFe LDHs as OER electrocatalysts have scarcely received a compact summarization on the aspect of unscrambling the intrinsically catalytic activity and enlightening the path for further development.
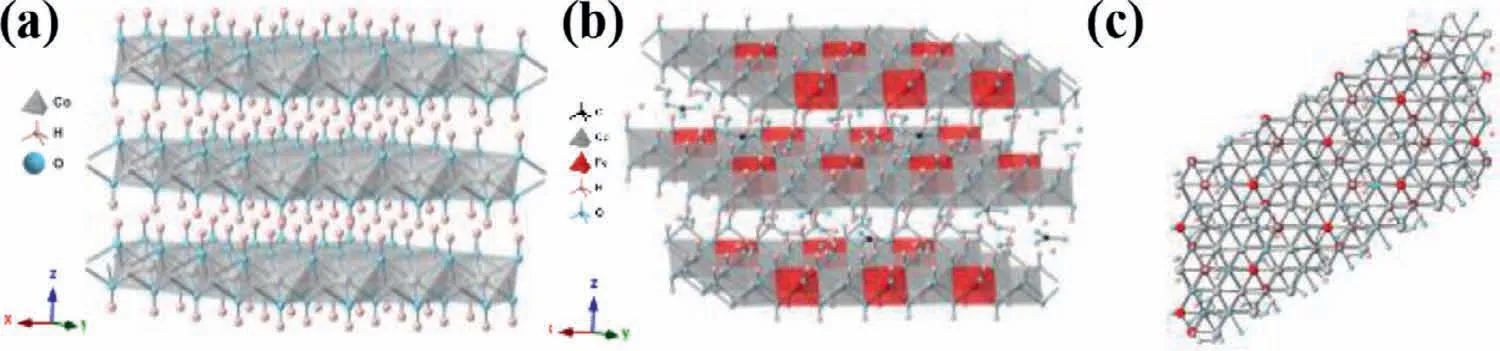
Fig.1.Ideal structure of (a) brucite-like β-Co(OH)2 and (b) hydrotalcite-like CoFe LDH (Co/Fe=3/1) stacked along the crystallographic c-axis with intercalated water and carbonate anions (randomly distributed).(c) Top-view along the c-axis of CoFe LDH.
Herein,we throw a concentrated attention on recent insights in CoFe LDHs for water oxidation,giving priority to their electronic configurations and chemical states which regulate the surface reconstruction for the generation of catalytically active phase and govern the binding energies of multistep OER intermediates as well.This review begins with the crystal structure of CoFe LDHs and the proposed OER mechanism,and then introduces the surface reconstruction and the key role of Fe species in CoFe LDHs,aiming at unveiling the structure-dependence of OER activity.Afterwards,representative optimization strategies for CoFe hydroxide catalysts are summarized and discussed thoroughly to point out the activity determinants at atomic level.Finally,we provide a concise outlook on the future development of CoFe LDHs for highperformance electrocatalytic water oxidation.
2.Fundamental of CoFe LDHs for OER
2.1.Crystal structure of β-Co(OH)2 and CoFe LDHs
In a nutshell,CoFe LDHs can be regarded as the substitution of iron for cobalt sites in the octahedron geometry of cobalt hydroxides and therefore it is convenient for us to use Co(OH)2to cotton on their crystallographic structures.The isostructuralα-Co(OH)2and stoichiometricβ-Co(OH)2phases are the two basic crystallographic forms of cobalt hydroxides.The former is a metastable phase constituted by positively charged Co(OH)2-xlayers with anions filled in the interlayer region for charge balancing,and facilely converts into aβ-Co(OH)2phase.The latter is composed of a hexagonal packing of hydroxyl ions with bivalent cobalt ions occupying alternate rows of octahedral sites (Fig.1),and Van der Waals forces exit between layers inβ-Co(OH)2,which is analogous to other layer materials (e.g.,graphite,MoS2).Brucite-likeβ-Co(OH)2
has a trigonal crystal symmetry with hexagonal lattice and space group P3m1.Fig.2 shows Bode scheme of squares for cobalt hydroxides and oxyhydroxide species.The phase transformation fromα-Co(OH)2intoβ-Co(OH)2is autonomous in thermodynamics and kinetics [25,26].Many factors can accelerate phase transformation,for instance,applied positive electric field,Fe impurities,intense stirring,etc.[26,27].The oxidation of Co(OH)2is much easier that than of Ni(OH)2,and the full oxidation of bivalent cobalt hydroxides are followed the redox transformation:
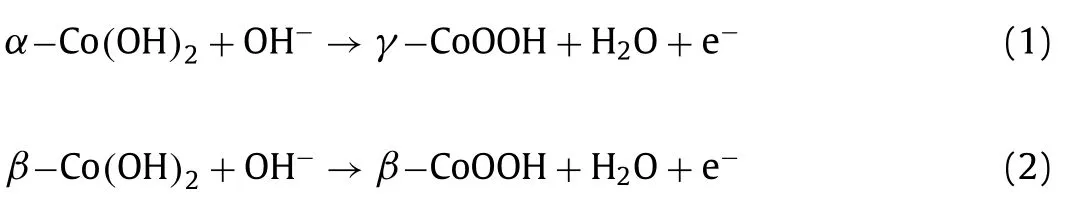
where the oxidation state of Co is+3 and+3/+4 inβ-CoOOH andγ-CoOOH,respectively.
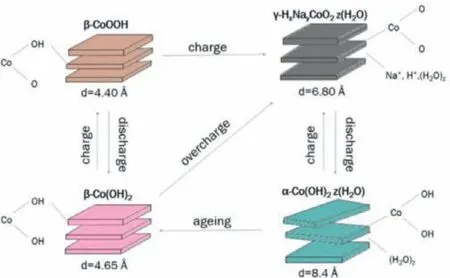
Fig.2.Bode scheme of squares for cobalt hydroxides and oxy-hydroxides species.Reproduced with permission [25].Copyright 2017,IOP Publishing.
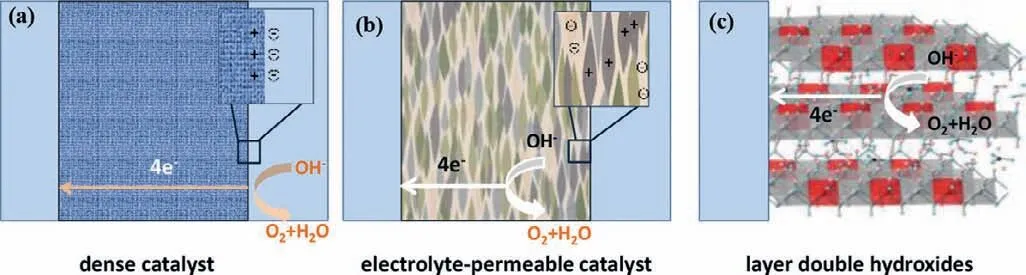
Fig.3.Comparison of dense,electrolyte-permeable and LDHs catalysts.(a) Dense catalysts are electrolyte-impermeable,and the surface charge of catalyst is compensated by an electrical double layer.(b) Electrolyte-permeable catalysts screen electronic charge on the catalyst with mobile solution ions (inset),leading to no electrostatic potential drop across the catalyst.(c) LDHs possess ionic conductivity and electrolyte intercalated between the nanosheets.
As for CoFe LDHs (Figs.1b and c),it can be obtained by the substitution of ferric iron for bivalent cobalt sites in octahedron geometry and the layers in Co(OH)2are resultantly positive charged.To balance these positive charged layers,anions (e.g.,CO32−,SO42−,Cl−,NO3−) intercalate in the interlayer region in cobalt hydroxides[17,24,28-32].However,the exchange of these interlayer anions with carbonate is unavoidable in alkaline electrolyte under ambient conditions and such anions exchange still occurs without an applied bias [33].Generally,CoFe LDHs can be expressed with the formula [17,28,34,35]:where x is the molar ratio of Fe in CoFe LDHs,A is the intercalated anion;n is the charge of the anions and y the amount of water between layers.
2.2.Proposed mechanisms for OER on CoFe LDHs
Prior to discussing OER mechanisms on (hydr)oxide surfaces,it is necessary to define the “surface” of (hydr)oxides where OER occurs because the intrinsic activity will be determined complicatedly [36,37].As shown in Fig.3,three possibilities have been put forward based on the degree of electrolyte permeability in electrocatalysts: (1) OER takes place at an outer surface,which is suitable for a dense catalyst with high degree of crystallinity.(2) Whole catalyst is OER active,which are appropriate for an electrolytepermeable catalyst,e.g.,amorphous hydroxides prepared under soft conditions.(3) OER occurs at a near surface region because of a specific structure of LDHs with ionic conductivity and electrolyte intercalating into the nanosheets [36–38].Therefore,layered CoFe LDHs provides more active sites for electrocatalytic water oxidation in contrast to oxide electrocatalysts of which OER takes place at an outer surface.However,the degree of electrolyte permeability in electrocatalysts greatly depends on the degree of crystallinity which is associated with preparation strategies.β-Co(OH)2and CoFe LDHs are generally prepared by soft methods (e.g.,precipitation methods and electrodeposition).Therefore,the hydrotalcitelike CoFe LDHs with low crystallinity are conceivably electrolytepermeable and therefore whole catalyst is OER active.The merit and demerit of electrolyte-permeable (amorphous phase and catalyst with low crystalline and high-disorder structure) and nonpermeable (high crystalline and dense structure) catalysts are summarized in Table 1.

Table 1 The physicochemical properties of amorphous and crystalline electrocatalysts.
There are several OER mechanisms of (hydr)oxides that have been proposed in recent decades.The classic adsorption mechanism is proposed that a battery of intermediates are chemisorbed and desorbed on the metal cations with concerted electron–proton transfers (CEPT),resulting in pH-independent activities.The release of oxygen molecule is not involved in lattice oxygen in catalysts.Whereas,lattice oxygen mechanism (LOM),which intensely depends on the co-valency of metal–oxygen bonds,is known as the release of oxygen molecule that is involved in redox reactions of lattice oxygen in catalysts and gives rise to pH-dependent activity[8].
As for CoFe,it is more suitable using the classic adsorption mechanism under alkaline conditions.The most critical and intricate step in OER multi-steps is the incipient O-O bond formation.One of these mechanisms can be expressed that O–O radical coupling interaction of metal-oxo or oxyl radical species (I2M)is involved in the formation of O-O bond Eqs.3-5.The alternative mechanism involves water/OH−nucleophilic attack (WNA) by a water molecule on an electrophilic MV–oxo or MIV–oxyl Eqs.6-9 [8,39].As shown in Fig.4,the involved OER multi-steps can be represented as follows:

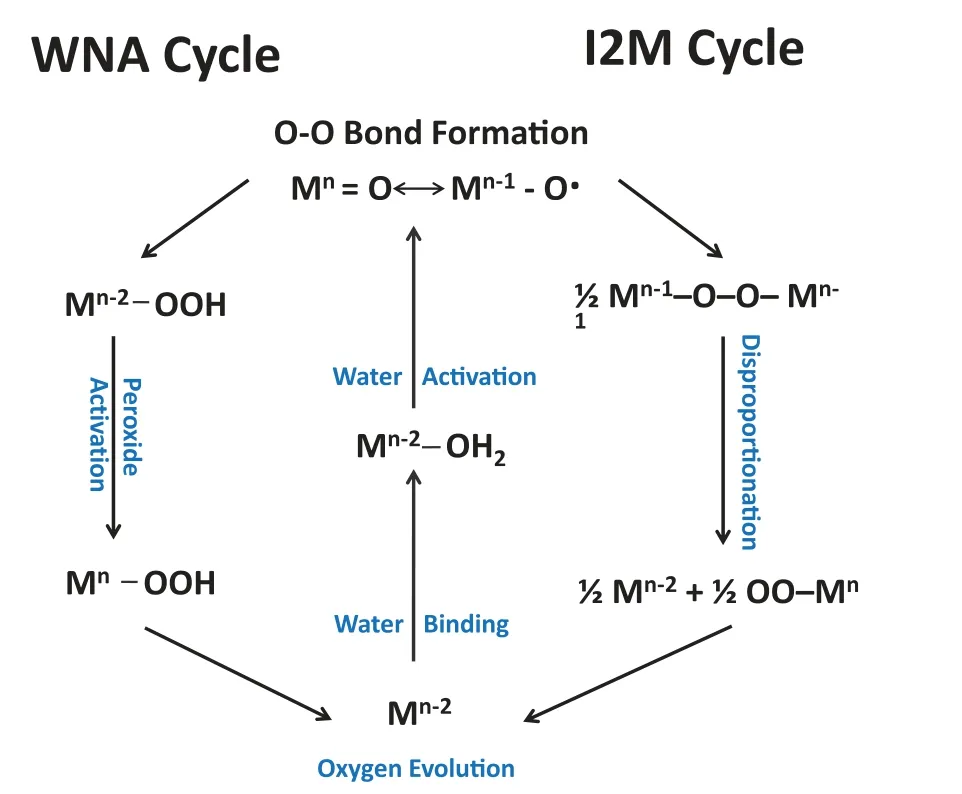
Fig.4.The proposed water nucleophilic attack (WNA) and oxo–oxo coupling (I2M)mechanisms for electrochemical water oxidation in alkaline conditions.
The M here represents the active sites of water oxidation catalysis.
Compared to the I2M cycle in water oxidation,the latter is more broadly used for theoretically modulating the process of OER.TheΔG1∼ΔG4denote as the four varied reaction free energies pertaining to a WNA cycle.Therefore,the potential determining step (PDS) rests with the largestΔGin the process of electrocatalytic OER and is conventionally the formation of MO from MOH or the evolution of MOOH from MO.Therefore,an OER activity descriptor is built on the difference betweenΔGO∗andΔGHO∗(ΔGO∗—ΔGHO∗) for the prediction of their OER performance and elucidate their intrinsic OER activity.According to the Sabatier principle,the adsorption strength of OER intermediates on an ideal catalyst is neither too strong nor too weak.The plot of overpotential as a function of (ΔGO∗—ΔGHO∗) results in the universal volcanoshaped relationship independent of electrocatalysts.
It is noteworthy that surface reconstruction and phase transformation in some case is mostly unavoidable for alkaline water electrolysis,even for hydroxides.Recently,operando Raman spectroscopy revealed that CoFe oxyhydroxide underwent a drastic reconstruction,spontaneously generating active CoOOH on its surface when immersed into KOH electrolyte,even under a zero bias condition [40].Dionigiet al.further demonstrated that the as-preparedα-phase CoFe LDHs was oxidized to the activatedγ-phase which was a feature of about 8% contraction of the lattice spacing and switching of the intercalated ions by using advanced electrochemical measurements,e.g.,operando X-ray scattering and absorption spectroscopy [41].However,the rational interpretations of crystal and electronic structures of active CoFe LDHs remain challenging during the OER process.Meanwhile,the synergistic effects between thein-situformed active phases and incipient substances still lack in-depth investigation.
2.3.The role of Fe sites in CoFe LDHs electrocatalysts
The most controversial issue in transition-metal based OER catalysts is the role of Fe spices because even trace amount of Fe impurities in electrolyte can obviously enhance pure Ni and Co based (oxy)hydroxides,whereas the catalytic performance of pure FeOOH is restricted by its extremely low electroconductivity [22,36,42,43].Enmanet al.manifested that the water oxidation activity of Co1-xFex(OOH) is about 100 times higher than pure CoOOH on a per-metal turnover frequency basis.Moreover,pure CoOOH is liable to be contaminated by Fe impurities using unpurified electrolyte.They proposed that CoOOH basically services as a conductive,high-surface area,and chemically stabilizing host for the most-active Fe sites [43,44].Operando X-ray absorption spectroscopy (XAS) has been employed in probing oxidation states,electronic structure,and local bonding environments of electrocatalysts under water oxidation condition,uncovering the nature of the catalytically active sites [45,46].As identified (Figs.5a-d),the iron was partially oxidized and the bond length of Fe-O was concomitantly shortened during water oxidation on Co(Fe)OxHy.The cobalt oxidation was only detected without the contamination of iron.These results demonstrated that CoOxHyfollows a varied water oxidation mechanism with and without iron corporation and suggested iron as active sites during water oxidation on Co-(Fe)OxHy[44].Different observations of the iron oxidation have been obtained and varied reaction mechanisms have been proposed.Gonget al.showed that CoOx,which adsorbed Fe3+ions from electrolyte,outperforms co-deposited Fe4.4Co95.6Oxfor water oxidation.Operando XAS manifested that the oxidation of Co sites in CoOx,CoOx+Fe3+,and control Fe48Co52Oxcatalysts was similar under water oxidation condition.But the adsorbed Fe3+with lower oxygen coordination on CoOxwas not further oxidized and the Fe-O bond lengths/strengths barely changed.The enhanced water oxidation activity of CoOxwas ascribed to the Fe sites with oxygen vacancies [47].Smithet al.observed the change of Co oxidation states of photochemically deposited Fe100-yCoyOxfilms,but not Fe under operando condition,confirming that OER takes place at two distinct reaction sites,di-μ-oxo bridged cobalt–cobalt and di-μ-oxo bridged iron–cobalt sites [48].
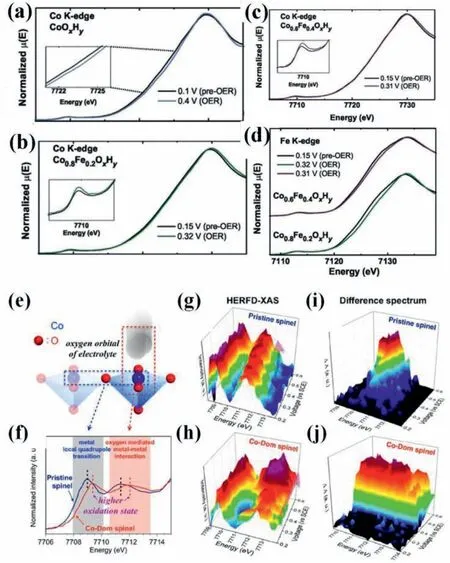
Fig.5.Operando Co K-edge XANES data collected for (a) CoOxHy,(b)Co0.8Fe0.2OxHy,and (c) Co0.6Fe0.4OxHy films.(d) Operando Fe K-edge XANES data.Pre-OER spectra were collected at potentials higher than the nominal Co2+/3+oxidation wave and prior to OER onset.OER spectra were collected at potentials necessary to maintain 3–4 mA/cm2 of OER current density.Potentials are given in overpotential,V vs.EO2/OH-.Reproduced with permission [44].Copyright 2018,Wiley-VCH.(e) Schematic illustration of the cobalt 3d orbital interaction of catalytic surface with oxygen 2p orbital of electrolyte.(f) High-energy resolution fluorescence detection X-ray absorption spectroscopy of Co K-edge for pristine spinel and Co-Dom spinel.Operando HERFD-XAS and difference spectrum of Co K-edge for(g,h) pristine spinel and (i,j) Co-Dom spinel.Reproduced with permission [21].Copyright 2018,American Chemical Society.
A synergistic effect of Co and Fe in CoFe LDHs for water oxidation,which is obligated to their superior OER activity,is proposed according to density functional theory (DFT) calculations.Dionigiet al.suggested that pure Co-Co centers and single Fe sites are less favorable for stabilizing OER intermediates than surface Fe sites which have synergy with nearest-neighbor Co sitesviaforming O-bridged Fe-Co reaction centers [41].Moreover,by taking advantage of operando high-energy resolution fluorescence-detected X-ray absorption spectroscopy (HERFD-XAS) with a small incident angle (Figs.5e-j),the more direct observation of local orbital interaction in reactant/catalyst interface can be achieved.The active sites of Co-Fe spinel oxides are Co ions rather than Fe,because O 2p orbitals directly interacted with 3d orbitals of Co ions.The key role of Fe ions is to stabilize the Co ions under a higher valent state during water oxidation and thereby may result in stable intermediate of reactant and its superior intrinsic activity [21].
These different features of Fe oxidation under OER condition were probably caused by the varied preparation of catalysts [44].However,we are not entirely sure whether Fe species participate in OER as active sites,according to the change of oxidation states of Fe species observed in a majority of operando XAS.This is because the orbital of oxygen will overlap with unoccupied Fe band under Fe4+condition,and this electron overlap with oxygen sites is the precondition by which discharge of oxygen atoms can occur with eventual release of O2molecules [22].Chunget al.proposed a dynamic reaction mechanism,which emphasized on balancing the rates of Fe dissolution and re-deposition over a MOxHyhost that constructed Fe active sites with dynamical stability.This is opposite to the traditional view featuring the confinement of all the components at the electrochemical interface (static active sites),and reckoning that only reactants and products are mobile [22].This is in line with the construction of dynamically stable Fe active sites in La1-xSrxCoO3model perovskite,which originate from the surface reconstructed thin Co hydr(oxy)oxide interacted with trace-level Fe impurities in the electrolyte.The dynamically stable Fe active sites strongly interacted with the Co hydr(oxy)oxide host endow with the OER activity of these perovskite catalysts[49].These observations and conclusions strongly strike the current OER mechanism and reaction descriptors (e.g.,orbital occupancy,metal–oxygen covalency and M-OH bond length),and will refine our understanding of OER reactions on 3d transition-metal hydr(oxy)oxides in alkaline media.
3.Synthetic methodologies
As shown in Fig.6,CoFe LDHs can be synthesizedviavarious protocols,including co-precipitation and homogeneous precipitation,electrodeposition and other alternative routes,thereby exhibiting different crystal sizes,long-range order and microstructures.The Gordian knot for the preparation of CoFe LDH is the appearance of unwanted phase (e.g.,FeOxHy),especially under circumstances with a lower atomic ratio of Co/Fe.
3.1.Co-precipitation and homogeneous precipitation methods
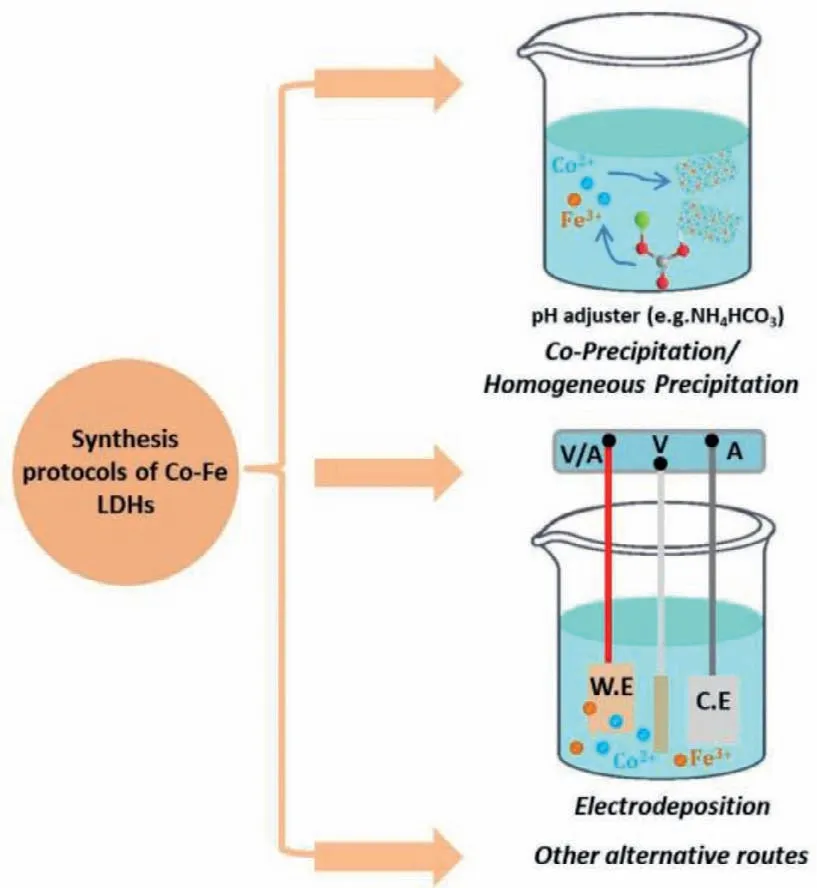
Fig.6.Representative synthesis protocols of CoFe LDHs: Coprecipitation/homogeneous precipitation,electrodeposition and other alternative routes.
The most ordinary methods for the synthesis of CoFe-based(oxy)hydroxides are co-precipitation and homogeneous precipitation,in which Co2+and Fe2+/3+salts and hydrolysis agents (e.g.,ammonia,sodium/potassium hydroxide,sodium/potassium carbonate,urea,hexamethylenetetramine) are dissolved in aqueous solution and react under heating and stirring.The morphological structure of CoFe LDHs is facilely controlled by the pH of deposition solution during nucleation.Elevating reaction temperature and prolonging time can increase the crystal sizes and improve crystallinity of CoFe LDHs [24].Hanet al.prepared CoFe LDHs nanosheets by a facile co-precipitation,which were further incubated at 80 °C for 48 h (hydrothermal method) to finally obtain carbonate-intercalated CoFe LDHs with high OER activity [50].
3.2.Electrodeposition
Electrodeposition is widely used in the preparation of materials due to its irreplaceable inherent merits [51,52].One of them is that the target materials can directly grow on conducting substrates(e.g.,carbon cloth,nickel foam,fluorine doped tin oxide),which ensures a low electron transfer resistance [51,52].A cathodic electrodeposition is generally conductedviathe cathodic reduction of NO3−or water at the conductive substrate surface,leading to the increase of pH and the consequential precipitation of CoFe LDHs[51].The plating solution contains bivalent cobalt and iron salts.The bivalent irons are facilely oxidized and become insoluble ferric hydroxide in the presence of air,and therefore solvent is usually purged with inert gases (N2and Ar) to get rid of dissolved oxygen prior to the dissolution of solutes [53,54].The only caveat in cathodic electrodeposition of CoFe LDHs is the different solubility product constant of bivalent cobalt hydroxide (1.09 × 10−15forαphase,5.92 × 10−15forβphase) and iron hydroxide (4 × 10−33),inducing different deposition speeds at electrode surface and consequently unexpected metal cation ratio in the same cases.
3.3.Other alternative routes
The alternative synthetic strategies for CoFe hydroxides are generally involved in the chemisorption of Fe impurity in electrolyte,a phase transformation from their solid-state phases into(oxy)hydroxides and a laser ablation in liquid (LAL) method [24].Due to the existence of Fe impurities in commercial KOH electrolyte,the Co (hydr)oxides inevitably chemisorb Fe impurities because of high affinity for the formation of CoFe hydroxide catalysts,and then dramatically enhanced the OER activity [9,22,43,44].For instance,Lopeset al.have proved that the OER activity of a La1−xSrxCoO3model perovskite originated from a thin surfacereconstructed Co hydr(oxy)oxide (CoOxHy) layer,and trace-level Fe species in electrolyte was dissolved and redeposited over CoOxHyhydr(oxy)oxide host,leading to the dynamically stable active sites[49].Gaoet al.synthesized CoO/CoFe LDHs hybrid catalystsviaa LAL method,in which a nanosecond pulsed laser beam was adopted to ablate a CoFe alloy (Co:Fe=3:1,atomic ratio) tablet immersed in NaCl aqueous solution [55].
4.Optimization strategies for CoFe LDHs electrocatalysts
As a traditional Chinese proverb “To do good work must first sharpen his tools” proves it,the profound understanding of the activity determinants is the prerequisite for the rational design of highly catalytic CoFe hydroxides.The OER activity of CoFe hydroxide catalysts depends on three aspects: (1) The number of active sites per unit volume.This is always ascribed to the electrochemical active area (ECSA),(or) specific surface area and the nature of catalysts whether electrolyte-accessible.(2) Electro-conductivity.In general,the strength of electronic conductivity is closely related to the number of co-valency in the M-Oπbond.Meanwhile,crystal defect is a critically key factor for enhancing the electronic conductivity of oxides.However,the charge transport in CoFe LDHs follows a mixed mechanism involving an “electron hopping” along the layers,which is caused by an inner redox reaction between oxidized and reduced forms of the Co2+/Co3+couple with a compensation of these extra positive charge by anions.Accordingly,under a suitable applied bias,the CoFe LDHs undergo redox reactions,prompting the charge transport among the CoFe LDHs [56].(3) The intrinsically catalytic activity of each metal cation in CoFe hydroxides.In recent years,several reaction descriptors have been proposed for the description of intrinsically catalytic activity of active sites,includingΔGO∗–ΔGHO∗,e.g.,orbital occupancy,metal–oxygen covalency,the binding strength of M-OH and Fe–M adsorption energy (M: Co,Fe,Ni,Cu,etc.) [8,22].Based on the above consensus,several approaches have been put forward to enhance the OER activity of CoFe LDHs relying on aforementioned four aspects,including nanostructured morphologies,doping foreign elements,defect engineering,hybridization,etc.
4.1.Nanostructured morphologies
The controllable synthesis of CoFe hydroxide catalysts with nanostructured morphologies is desirable due to their high specific surface area.As OER occurs at the surface of catalysts,the increase in surface area is certainly conducive to enhancing water oxidation efficiency.This is because larger surface area of electrocatalysts can afford more active sites,shorten charge transport distance and facilitate mass transfer [57].Moreover,CoFe hydroxide catalysts with nanostructured morphologies potentially enrich more crystal defects,further facilitating the OER activity [58].In addition,due to the layered structure,few-layered and mono-layered CoFe LDHs electrocatalysts can be obtainedviaexfoliation of the raw materials or direct preparation under suitable condition,which gives rise to an ample amount of active sites,convertible electron configuration,and more distorted and defective structures.For instance,Zhouet al.fabricated a single-layer CoFe LDHs under a near-anhydrous condition.Owing to increasedCdl,more crystal vacancies,larger degree of structural disorder and enhanced intrinsic activity,single-layer CoFe LDHs outperformed bulk materials for water oxidation and afforded overpotential of 270 mV at 10 mA/cm2[59].CoFe LDHs are facilely exfoliated into ultrathin structure with abundant vacancies.This will be clarified in section 4.3.Recently,DFT calculations demonstrated that the edge (100) facet of NiFe based electrocatalysts exhibits a much higher OER activity than basal plane (001) facet [60,61].Due to the similar layered structure,the exposed edges sites on (100) of CoFe LDHs are expected to have a higher intrinsic activity.Therefore,the OER performance of CoFe LDHs is supposed to be likely enhanced by increasing the ratio of the exposed edges sites,while there is no direct evidence for enhancing water oxidationviaaforesaid optimizing strategy at the present stage.
CoFe LDHs can also be used as precursors for the preparation of CoFe oxides,selenides,sulfides and alloy,which maintains similar morphologies [28].For instance,Liet al.synthesized 2D CoFe alloy nanosheet arrays with atomic thickness on Ni foamviaa topotactic reduction of CoFe LDHs [62].
4.2.Doping foreign elements
Doping foreign elements is a conventional and effective approach to boost the OER activity by increasing their intrinsic activity of active sites and (or) prompting the creation of catalytically active phase [13,24,63].The increased electrical conductivity,changed electronic structure and accelerated surface reconstruction of catalystsviaforeign elements substitution can be straightforwardly understood by virtue of molecular orbital theory and chemical/physical characterizations.
Vanadium (V),tungsten (W),chromium (Cr) and cerium (Ce)with high valence state and highly electronegative fluorine (F) are the most hackneyed dopants for enhancing OER activity by drawing or pulling the electron from neighboring 3d metals and therefor optimizing the binding energies of the intermediates [64–71].Huet al.found Co and Fe 2p in CoFe LDHs exhibit various degrees of positive shift after doping high valence state V and show an intense electron correlation in Co,Fe and V,accounting for the boosted OER activity [65].Moreover,Yanget al.considered that V doping can improve the covalency of the metal-oxygen bond because of slightly blue shift of binding energy of Co,Fe and O and the consequent charge-transfer from oxygen to the metal for OER[72].By using CHF3-plasma etching technique,Liuet al.prepared F-doped Co3Fe LDH ultrathin nanosheets with an excellent OER activity due to the filling of oxygen vacancies with F−ions and decreasing the reaction energy barrier [66].In some cases,noble metal cations have also been implanted into transition-metal based catalysts for lowering the amount of noble metals [73,74].For instance,Zhuet al.found Rh doping in CoFe LDH would create Fe vacancies,which led to the changes in the coordination environment of Fe.The special character of Rh doping and their hollow structure promoted whole HER and OER processes [73].
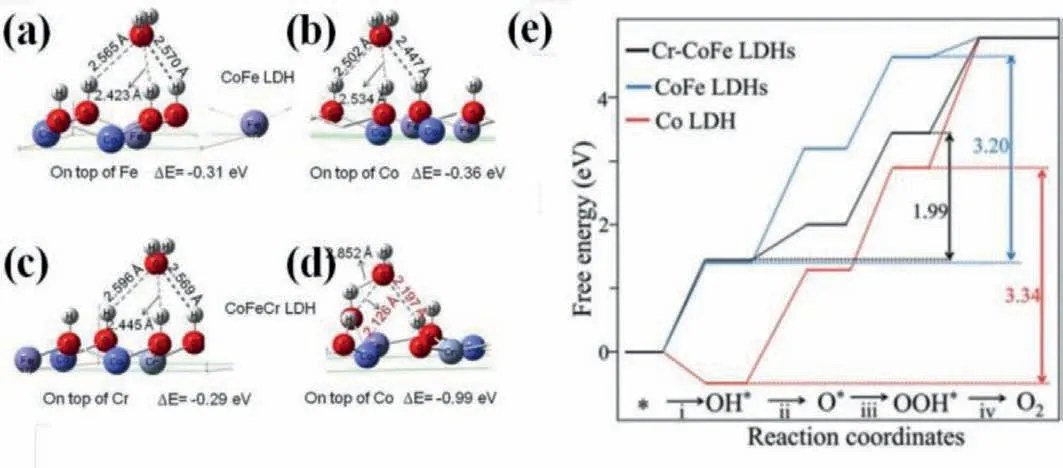
Fig.7.Adsorption energies for H2O on different sites.On top of (a) Fe and (b) Co sites of CoFe LDH;On top of (c) Cr and (d) Co sites of CoFeCr LDH.Reproduced with permission [64].Copyright 2020,Elsevier.(e) Free energy diagram for the OER on three considered models at zero potential.Reproduced with permission [67].Copyright 2019,Wiley-VCH.
The improved intrinsic activity of active sitesviadoping foreign elements can be expressed by the Gibbs free energy changes in catalysts because OER reaction pathways with multi-step intermediates can be simulated with the aid of first principles calculations based on DFT [13,17,57].For instance,doping foreign elements can enhance the adsorption of H2O molecules on active sites,which is deemed as a determining step and can be applied for the estimation of active sites and catalytic activity.As shown in Figs.7a-d,the pristine adsorption energies for H2O molecules on Fe and Co site are –0.31 and –0.36 eV,respectively,indicating Co atom possesses superior catalytic activity and acts as active sites for water oxidation.Whereas,adsorption energies for H2O molecules on Co site can be reduced into –0.99 eV after Cr-doping,much lower than those on Cr atom (–0.29 eV) and pristine Co site.Moreover,the reduced adsorption energies for H2O on Co atom is coincide with the decreased bond lengths which derives from the interaction between oxygen atom in water molecule and surface H atom in catalyst [64].Therefore,Cr doped CoFe LDHs affords ultralow overpotential of 202 mV at 10 mA/cm2(Table 2).Doping foreign elements,especially iron ions,in other first-row transition metal based catalysts can alter the PDS,switch the active sites from other first-row transition metal to dopants and decrease the overpotential for water oxidation [22,44,49].As shown in Fig.7e,the PDS on Co LDHs is the formation of oxygen molecule and the calculated overpotential is 0.81 eV,which reduces to 0.55 VviaFe incorporation along with the formation of∗O from∗OH being the PDS.However,the multi-step intermediates during the OER process were chemisorbed on Cr sites in Cr-CoFe LDHs rather conventional Co sites,leading to similar adsorption strength of OH on the Cr-CoFe LDHs (ΔEOH=1.06 eV) and CoFe LDHs (ΔEOH=1.06 eV)and decreased adsorption strength toward OOH∗from 3.06 eV for Cr-CoFe LDHs to 4.25 eV for CoFe LDHs.Besides,the highest energy barrier is the evolution of oxygen molecules from ∗OOH with overpotential of 0.23 V on Cr-CoFe LDHs,which is much smaller than that on CoFe and Co LDHs [67].
Nickel foam (NF),carbon paper (CP) and glassy carbon electrode (GCE) are commonly used as supports.The current density(μA/cm2ox) was normalized to the surface area of the oxides to give the intrinsic activity.Surface reconstruction has attracted attentions for in-depth understanding of structure-activity relationship of catalysts.Typically,under a high anodic oxidation condition,the surface of catalysts suffer some degree of reconstruction for the generation of a thin (oxy)hydroxide layer which is the truly active phase for water oxidation [75].Doping foreign elements can facilitate this process and regulate the electronic configurations of reconstructed oxyhydroxides,thus promoting the OER activity.Recently,spinel CoAl2O4with OER inactivity can be intensely activated by Fe substitution,owing to the facilitated reconstruction into Co oxyhydroxides [63].Meanwhile,Fe substitution leads to the oxygen being negatively charged due to the activation of deprotonation on the reconstructed oxyhydroxides and thereby oxygen can act as the reactive site for oxygen evolution.Therefore,Fe doping remarkably enhances the water oxidation performance of CoAl2O4,which outperforms the benchmark IrO2.Besides,Co is promoted to be pre-oxidized and the structure of Co2Al2O4processes great flexibility because Fe substitution propels the upshift of oxygen 2p levels.Accordingly,oxygen vacancies are accumulated at the surface along with lattice oxygen oxidation that ends at the dissolution of Al3+,maintaining a stable surface chemistry [63].The similar surface reconstruction on LiCoO2electrocatalystviaCl doping has been also discovered.Wanget al.carried out a cationic redox-tuning strategy to regulatein-situcatalyst leaching and to redirect the dynamic surface restructuring of layered LiCoO2–xClx(x=0,0.1 or 0.2),for OER.LiCoO1.8Cl0.2required lower potential to initiatein-situCo oxidation and Li leaching in contrast with Cl-free LiCoO2,resulting in surface reconstruction for the generation of self-terminated amorphous (oxy)hydroxide during the OER.Moreover,the stabilization of surface reconstruction on LiCoO1.8Cl0.2called for shorter cycles than that of Cl-free LiCoO2.Surface-restructured LiCoO1.8Cl0.2afforded a superior catalytic activity among many state-of-the-art OER catalysts and showed significant stability [76].The OER performance of representative catalystsviadoping foreign elements is summarized in Table 2.
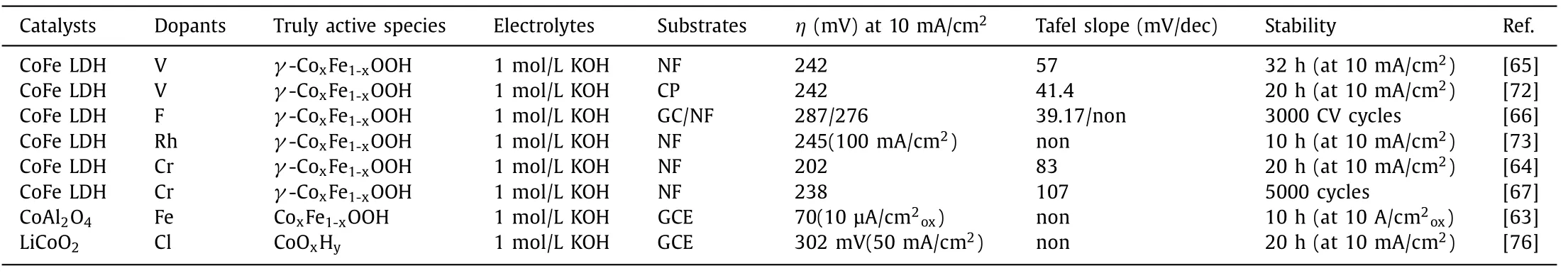
Table 2 OER performance of representative catalysts via doping foreign elements.
In spite of the glaring research on the mechanism of doping,yet,there are still some dark spots in the bright sky.V,W and chromium Cr based (hydr)oxides are amphoteric compounds and facilely dissolved in alkali electrolyte,especially under high oxidation conditions.More seriously,surface reconstruction of electrocatalyst is inevitable because oxyhydroxides are conceived as truly catalytically active phase [75].Hence,do aforementioned dopants still exit in the reconstructed amorphous oxyhydroxides? If not,how to explain improved OER activity of catalysts? Creating vacancies,promoting surface reconstruction and (or) facilitating the adsorption/interaction of Fe impurities [49,77-79]? Particularly,we might be more cautious for the effect of Fe impurities in commercial electrolyte because of recent works put emphasis on dynamically stable Fe active sites in La1-xSrxCoO3model perovskite whose OER activity originated from the surface reconstructed thin Co hydr(oxy)oxide interacted with trace-level Fe impurities in the electrolyte [49].
4.3.Defect engineering
The existence of crystal defects in materials is not preventable,and a moderate amount of defects can improve their electroconductivity,tune electronic structure and provide more unsaturated cation sites for electrolysis.This is because oxygen surpluses or vacancies induce charge counterbalancing holes or electrons in the metal d orbital manifolds [80]and OH−is firstly chemisorbed on the surface O vacancy site [8].Therefore,defect engineering is conceived as a feasible strategy for promoting OER activity.The most common strategies for creating oxygen vacancies in targeted catalysts are the utilization of reductive agents (TiCl3,NaBH4,etc.)and gases (H2,NH3,CH4,etc.).The implantation of electrochemical reduction is also feasible to create oxygen vacancies [81].But it is more difficult and complicated for the introduction of cation defects in solid catalysts,relying on special formation mechanism[82,83].As for layered hydroxides,one of the most conventional methods is to exfoliate the bulk materials into and (or) directly fabricate ultra-thin nanosheets with atomic thickness,thus simultaneously creating defects and increasing the number of active sites for OER [17,28].For instance,Zhouet al.proposed a simple co-precipitation method for the synthesis of single-unit-cell-thick and defect-rich LDH nanosheets [84].However,it is worth noting that the re-stacking of few-layered and mono-layered CoFe LDHs is conceivably inevitable after a series of post-processing (e.g.,centrifugation and drying).Therefore,the synthetic strategies centering on the synthesis of CoFe LDHs nanosheets with atomic thickness on conductive substrate (e.g.,nickel foam,carbon cloth,Cu foils,Ti mesh) are more desirable and suitable.Besides,the exact layer number control from monolayer to multilayer (e.g.,1 →5)remains intriguing but challenging.Advanced synthetic methodologies should be carried out for achieving these arduous targets.
Plasma etching has been widely implemented in the introduction of vacancies in solid catalysts [72,73,85].Liuet al.proposed water-plasma-enabled exfoliation of CoFe LDHs nanosheets along with producing multi-vacancies due to the destruction of the electrostatic interactions between the host metal layers and the interlayer cations (Fig.8a).The applied potential arriving at 10 mA/cm2of LDHs is reduced from 1.561 into 1.520vs.RHE (Fig.8b).The improved OER activity of CoFe LDHs is consequently derived from the increment in active sites and multi-vacancies [20].More critically,N2,Ar and CHF3-plasma etching have also been applied to create vacancies in CoFe LDHs and give rise to a significantly enhanced electrocatalytic activity for OER [18,19,66].Recently,Liuet al.manifested a synergistic effect of Ce doping and plasma etching in CoFe LDHs (V-Ce/CoFe LDHs) for enhanced HER (Fig.8c).The plasma etching brings about massive defects and triggers amorphous phase formation in CoFe LDH,meanwhile,Ce doping results in additional active sites and improves its electrical conductivity[71].Therefore,V-Ce/CoFe LDHs deliver remarkably small overpotential at 10 mA/cm2(73 mV) for HER (Fig.8d).Besides,as shown in Figs.8e and f,Dinget al.found the OER activity of ultrathin CoFe LDHs nanosheets obtained by Ar-plasma exfoliation of bulk counterparts is harvested under illumination due to photoelectronic excitation and oxo-bridge-mediated metal-to-metal charge-transfer transition [86].Yanget al.provided other alternative strategies for inducing vacancies in (hydr)oxides by facile H2O2treatment.The Fenton reaction occurs between the tervalent Fe and H2O2.Accordingly,the generated oxygen and oxygen free radicals reduced integrant Fe3+into Fe2+along with the oxidation of Co2+,leading to multiple defects in CoFe hydroxides [87].The OER performance of representative catalystsviadefect engineering is summarized in Table 3.
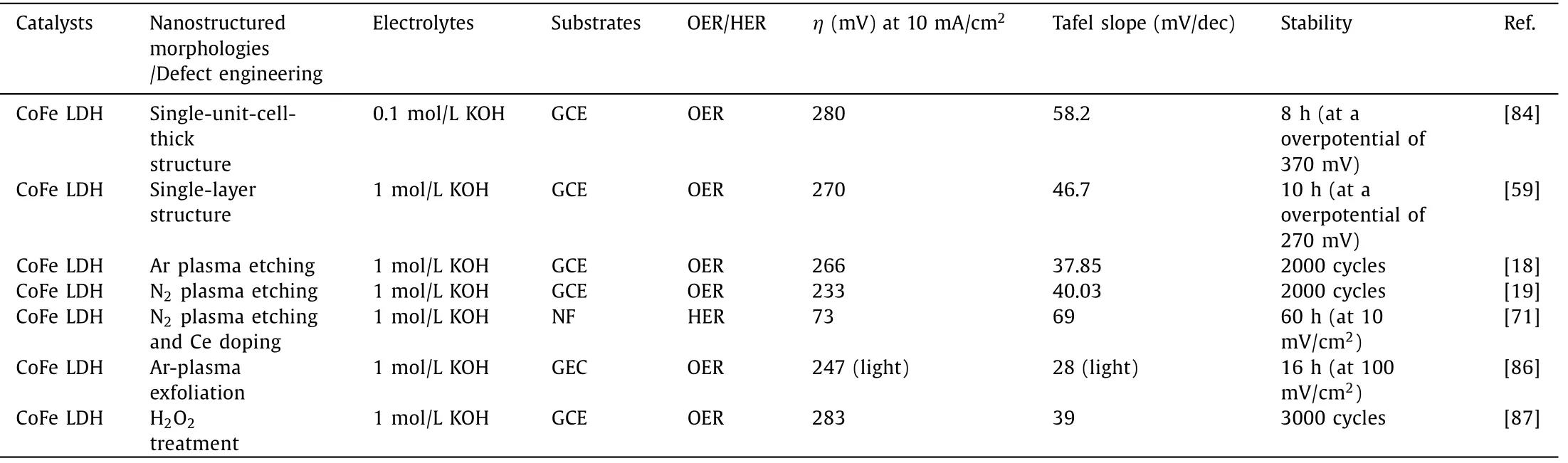
Table 3 OER performance of representative catalysts via defect engineering.
4.4.Hybridization
In broad terms,anchoring the targeted catalysts on other disparate materials forms hybrid catalysts,leading to increased electrolyte-accessible sites,enhanced electron transport and tunable local chemical binding environment of hybrid interfaces [88–93].Hybrid catalysts usually take on multiple forms,including catalyst@conducting substrate [89,94],catalyst@support [95–102],and catalyst@catalyst [55,103-105].The glassy carbon electrode (GCE),Au substrate,titanium mesh,Ni foam (NF),carbon paper (CP) and carbon cloth (CC) are the most conventional substrates,while supports are of high diversity,including various types of carbon materials [24,91,93],metallic copper and C3N4semiconductor (Figs.9a and b) [101,102],etc.However,the rational interpretation of local chemical binding environment of hybrid interfaces and the corresponding catalytic mechanisms,especially for catalyst@catalyst hybrids,is desirable but challenging.
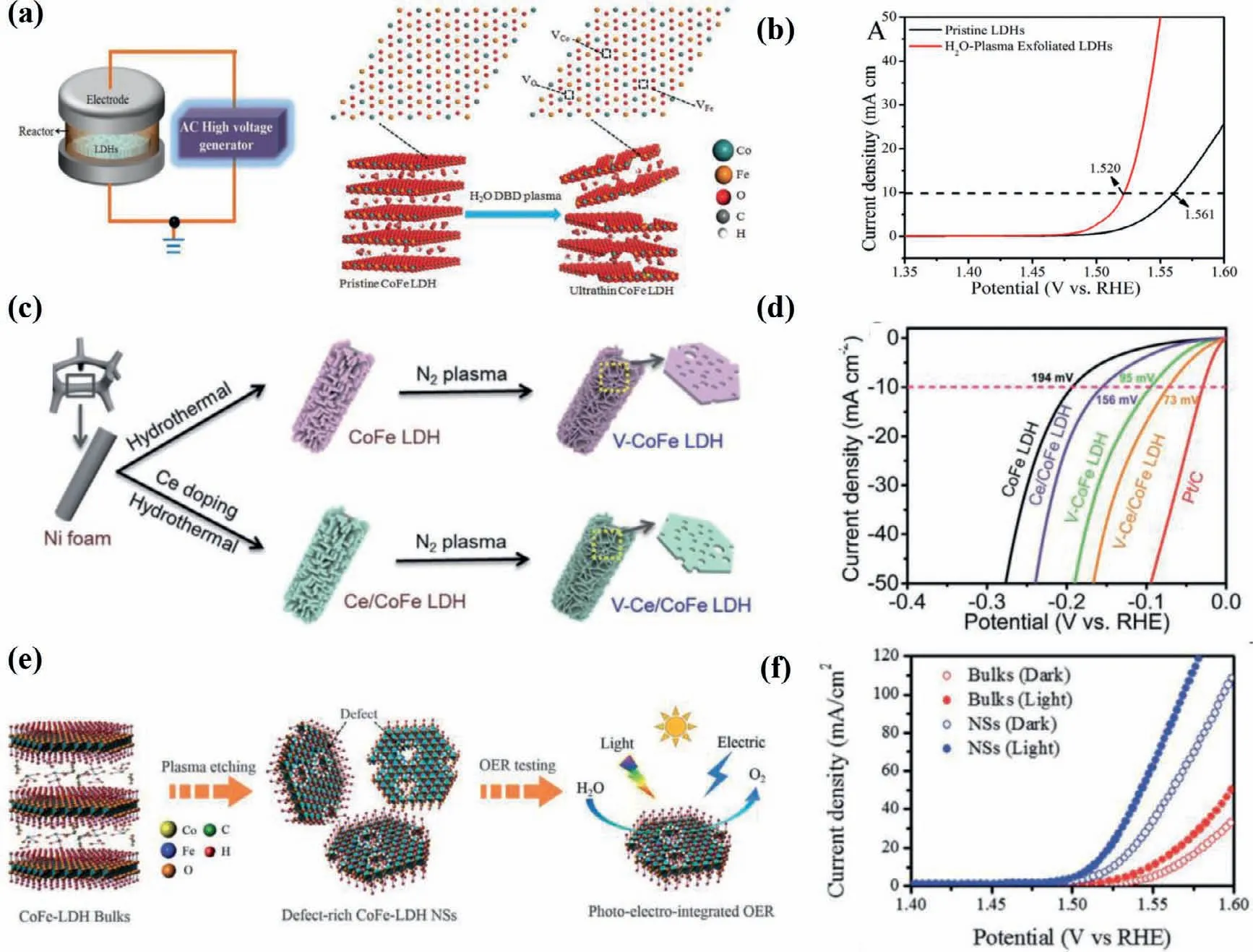
Fig.8.(a) Schematic illustration of the water-plasma-enabled exfoliation of CoFe LDH nanosheets.The DBD plasma reactor is designed with the plate-to-plate electrode at 50 V powered by the AC high voltage generator.Reproduced with permission [20].Copyright 2017,Wiley-VCH.(b) LSV curves for OER on pristine CoFe LDHs and the waterplasma exfoliated CoFe LDH nanosheets.(c) Schematic illustration of synthesis of V-CoFe LDH and V-Ce/CoFe LDH.HER performances of CoFe LDH,V-CoFe LDH,Ce/CoFe LDH and V-Ce/CoFe LDH in 1 mol/L KOH electrolyte: (d) IR-corrected polarization curves.Reproduced with permission [71].Copyright 2020,The Royal Society of Chemistry.(e) Schematic illustration of the Ar-plasma etching of bulk CoFe LDHs into defect-rich ultrathin nanosheets (NSs) to highlight photo-electro-integrated OER activity.(f) OER polarization curves of CoFe-LDH bulks and NSs in dark and light fields.Reproduced with permission [86].Copyright 2020,The Royal Society of Chemistry
To boost OER activity,conducting substrates and supports with superior conductivity and high specific surface area,give rise to faster mass transfer kinetics,more exposed active sites and higher bubbles evolve rates at the electrodes under operation condition.It is noteworthy that fast bubbles evolve rate at the electrodes surmounts the bubble overpotentials and avoids catalytic deactivation,maintaining OER stability of catalysts [24].Wanget al.electrodeposited CoFe LDHs on CuO nanowire array,forming a CuO@CoFe LDHs core−shell heterostructure,which exhibited highly catalytic activity and excellent stability toward OER under mild alkaline conditions.In contrast to the first two hybrids,the catalyst@catalyst hybrids are hackneyed but tanglesome hybrid category because electrochemical reaction presumably occurs at the whole hybrids,affording luxurious active sites.As shown in Fig.9c,Huanget al.proposed a hierarchical FeCo2S4@CoFe LDH core–shell structure catalyst by a hydrothermal–sulfuration–electrodeposition three-step strategies.The special structure of FeCo2S4@CoFe LDHs hybrids enriches abundant catalytically active sites,leading to small overpotentials of 115 and 247 mV to reach a current density of 10 mA/cm2for the HER and OER,respectively [104].Meanwhile,the support also affects the direct growth orientation of catalyst and alters the physicochemical property of catalyst.In particular,Haoet al.found the hydroxyl-group on MXene facilitated surface growth and maximized the exposed active edge sites of the CoFe LDHs,and the O 2p states in LDHs become distributed above the Fermi level which is mediated by the possible anionic redox process,suggesting the insulated LDH possesses metallic features(Fig.9d-f) [106].More crucially,the existence of intense chemical couplings at the interface of hybrids can promote the charge transfer between hybrids and alter the electronic structure of metal cations,affecting the water oxidation efficiencies.For instance,Maet al.conceived that the intense interfacial coupling effects between CoFe LDHs and Co3O4are conducive to their superior OER activity and faster charge transfer than individual component [103].As shown in Fig.9g,Gaoet al.found that trigged by the intense chemical couplings at the interface of hybrids,the charges transfer from bivalent Co in CoO to trivalent Fe in the CoFe LDHs through the interfacial Fe-O-Co bond,enabling more Co3+sites with higher valence state in the hybrids [55].The OER performance of representative catalystsviahybridization is summarized in Table 4.
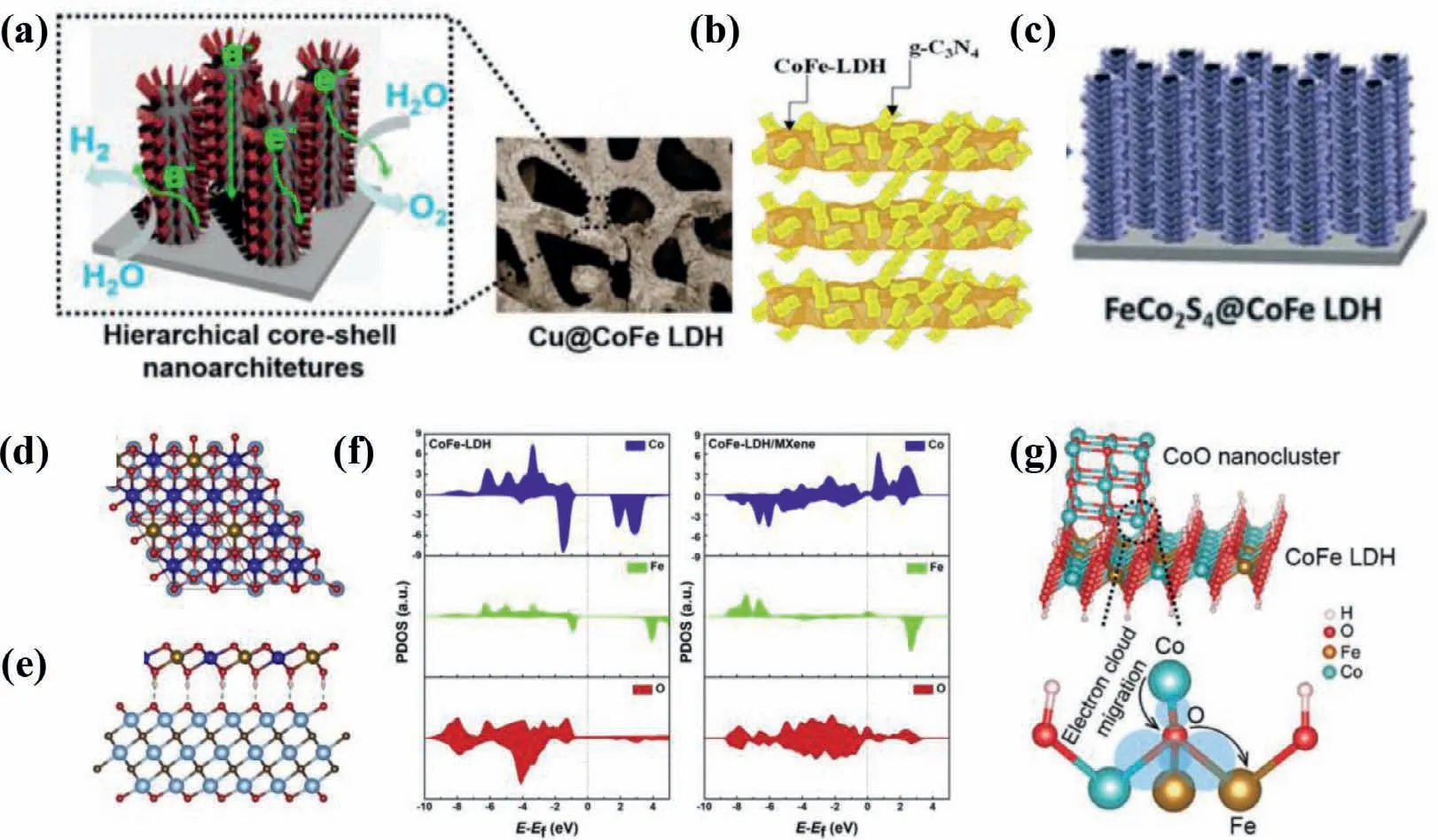
Fig.9.(a) Cu@CoFe LDH core-shell nanostructure electrocatalysts.Reproduced with permission [102].Copyright 2017,Elsevier.(b) CoFe-LDH@g-C3N4 hybrid semiconductor.Reproduced with permission [101].Copyright 2019,The Royal Society of Chemistry.(c) FeCo2S4@CoFe LDH hybrid.Reproduced with permission [104].Copyright 2020,The Royal Society of Chemistry.(d) Top view and (e) side view of model structure of CoFe-LDH/MXene hybrid system.The Co,Fe,Ti,O,C and H atoms are presented in blue,yellow,light blue,red,grey and white colors,respectively.(f) Projected density of states (PDOS) of individual CoFe-LDH and CoFe-LDH/MXene.The Fermi level is shifted to zero.Reproduced with permission [106].Copyright 2019,Elsevier.(g) Schematic illustration of electron transfer at the interface of the M-CoO/CoFe LDHs hybrid.Reproduced with permission [55].Copyright 2018,Wiley-VCH.
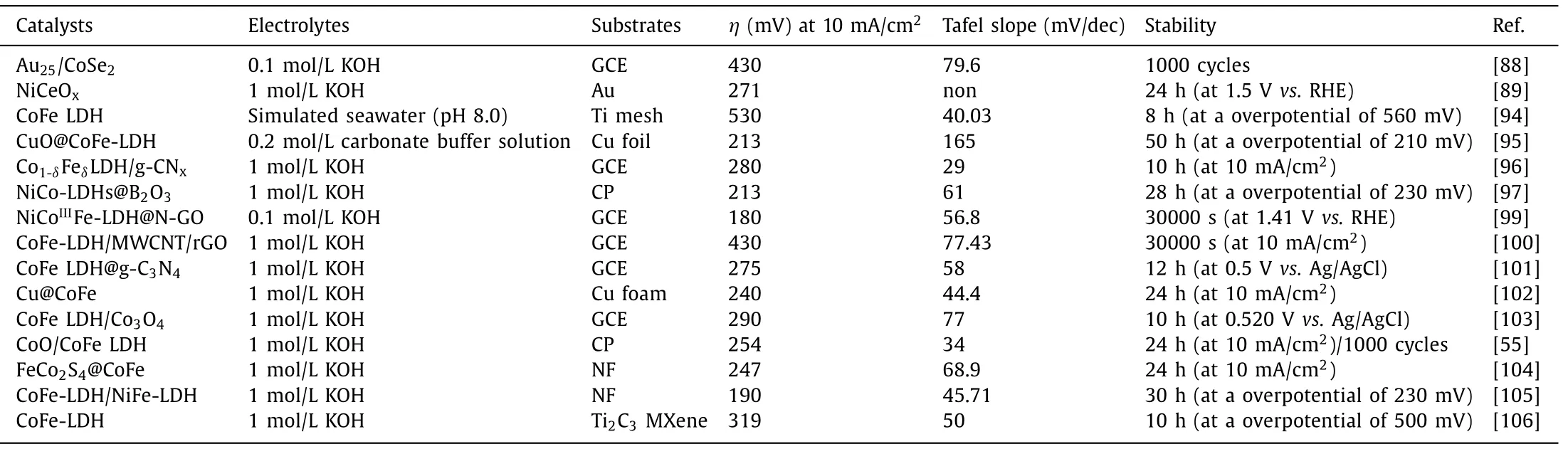
Table 4 OER performance of representative catalysts via hybridization.
5.Summary and outlook
CoFe-based (oxy)hydroxides have been conceived as alluring OER electrocatalysts because of high catalytic activity and superior endurance against the corrosion under alkaline condition,surpassing many kinds of the state-of-the-art noble metal based catalysts.In this review,the in-depth understanding of crystal structure and phase transformation of CoFe hydroxides has been achieved,uncovering the structure-activity relationship.Prior to understand the basic OER mechanism,we firstly discussed the “surface” of(hydr)oxides that OER occurs for the better understanding intrinsically catalytic activity of catalysts.In quick succession,we discussed the key role of Fe species and analysis of surface reconstruction/phase transformation of CoFe LDH during OER.Advanced optimization strategies for CoFe hydroxide catalysts have been summarized centering on enhancing electro-conductivity,enlarging the active sites and regulating the electronic structure to optimize the binding energy of the intermediates.
At the present stage,the most urgent topic is the design of high efficient CoFe LDHs based electrocatalyst with superior stability at atomic level,satisfying the technical requirements of liquid sunshine visions and other commercial utilization.There is the dispute about the active sites in CoFe LDHs for water oxidation,which blocks probing OER mechanism.The significant impacts of Fe impurities are non-negligible,and therefore excluding the impact of Fe impurities in electrolyte is prerequisite for asserting the mechanism of improved OER.More critically,CoFe LDHs oxidize from as-preparedα-phases to activatedγ-phases prior to the process of water oxidation.At first,the interpretation of crystal structure and electronic structure of this activatedγ-phases is still intriguing but elusive.Moreover,it is unclear whether there is a synergistic effect between thein-situformed active phase and incipient substances.Therefore,advancedin-situ/operando techniques along with theoretical predictions and modeling should be employed for wrestling with aforesaid enigma of CoFe LDHs during OER process.In addition,CoFe LDHs often suffer deactivation for water oxidation,leading to an inferior long-term stability.The corresponding deactivation mechanisms and protecting strategies should be put forward.For instance,the porous substrates and supports can be used for avoiding bubble overpotentials and resultant detachment of catalyst from the electrodes.A proper amount of Fe impurities can be intentionally added into electrolyte,giving rise to a dynamical balance of dissolution and re-deposition of Fe species over a CoOxHy.Finally,more attention should be paid to designing advanced water electrolysis reactors and devices for increasing the collection,separation and storage efficiencies of hydrogen.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos.21773093 and 22175077),Natural Science Foundation of Guangdong Province (Nos.2021A1515012351 and 2017B030306004) and Guangdong Special Support Program (No.2017TQ04N224).
 Chinese Chemical Letters2022年6期
Chinese Chemical Letters2022年6期
- Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria