
Synthesis and characterization of the two enantiomers of a chiral sigma-1 receptor radioligand: (S)-(+)- and (R)-(-)-[18F]FBFP
2022-07-09 02:14ToWngYingZhngXiojunZhngLyunChnMingqingZhngJinmingZhngPtrBrustWinniDuthrConrYiyunHungHongmiJi
Chinese Chemical Letters 2022年7期
To Wng, Ying Zhng, Xiojun Zhng, Lyun Chn, Mingqing Zhng,Jinming Zhng,*, Ptr Brust, Winni Duthr-Conr, Yiyun Hung,*, Hongmi Ji,*
a Key Laboratory of Radiopharmaceuticals (Beijing Normal University), Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China
b Nuclear Medicine Department, The First Medical Center of Chinese PLA General Hospital, Beijing 100853, China
c Institute of Radiation Medicine, Peking Union Medical College & Chinese Academy of Medical Sciences, Tianjin 300192, China
d Yale PET Center, Department of Radiology and Biomedical Imaging, Yale University School of Medicine, New Haven, CT 06520-8048, United States
e Helmholtz-Zentrum Dresden-Rossendorf, Institute of Radiopharmaceutical Cancer Research, Department of Neuroradiopharmaceuticals, Leipzig 04318,Germany
ABSTRACT Racemic [18F]FBFP ([18F]1) proved to be a potent σ1 receptor radiotracer with superior imaging properties.The pure enantiomers of unlabeled compounds (S)- and (R)-1 and the corresponding iodonium ylide precursors were synthesized and characterized.The two enantiomers (S)-1 and (R)-1 exhibited comparable high affinity for σ1 receptors and selectivity over σ2 receptors.The Ca2+ fluorescence assay indicated that (R)-1 behaved as an antagonist and (S)-1 as an agonist for σ1 receptors.The 18F-labeled enantiomers (S)- and (R)-[18F]1 were obtained in >99%enantiomeric purity from the corresponding enantiopure iodonium ylide precursors with radiochemical yield of 24.4% ± 2.6% and molar activity of 86–214 GBq/μmol.In ICR mice both (S)- and (R)-[18F]1 displayed comparable high brain uptake, brain-to-blood ratio, in vivo stability and binding specificity in the brain and peripheral organs.In micro-positron emission tomography (PET) imaging studies in rats,(S)-[18F]1 exhibited faster clearance from the brain than (R)-[18F]1, indicating different brain kinetics of the two enantiomers.Both (S)- and (R)-[18F]1 warrant further evaluation in primates to translate a single enantiomer with more suitable kinetics for imaging the σ1 receptors in humans.
Keywords:σ1 receptor Enantiomer Radiotracer Positron emission tomography Fluorine-18
The sigma-1 (σ1) receptor consists of 223 amino acid residues[1] with molecular weight of 25.3 kDa [2].It is mainly localized at the endoplasmic reticulum (ER) membrane, specifically mitochondria-associated membrane (MAM) [3,4].Most importantly, this receptor is a unique “ligand-operated receptor chaperone” and interacts with many functional proteins [4].The crystal structure of humanσ1receptor revealed a trimeric architecture with a single transmembrane domain in each protomer [3].It can be modulated by drugs and differences between agonistand antagonist-bound crystal structures of theσ1receptor have been illustrated recently [5].Agonists decrease whereas antagonists increase the oligomeric state ofσ1receptor [6–8], suggesting that agonists and antagonists dynamically regulateσ1receptor oligomerization in distinct manners [9].
Theσ1receptor has been shown to regulate various physiological processes, such as transcriptional activity, Ca2+homeostasis, ER stress response, and autophagy [10–12] with its role in organizing and remodeling cholesterol-enriched ER signaling microdomains [13].Growing evidence demonstrated that this chaperone is also involved in a number of disorders including central nervous system (CNS) diseases [14–21], cardiovascular diseases [22] and cancers [23].In the CNS, theσ1receptor plays a pivotal role in the pathophysiology of many neuropsychiatric disorders including Alzheimer’s disease (AD), Parkinson’s disease(PD), Huntington’s disease (HD), frontotemporal lobe degeneration(FTLD), amyotrophic lateral sclerosis (ALS) and major depressive disorder (MDD) [14–21].Several compounds acting on theσ1receptor including agonists such as ANAVEX2–73, SA4503 and pridopidine and antagonists S1RA have entered clinical trials as drug candidates, underscoring the importance ofσ1receptor as a therapeutic target [19,24].Most recently, theσ1receptor has been identified as an essential host factor supporting SARS-CoV-2 viral infectivity [25,26].The availability of an optimal radioligand for positron emission tomography (PET) imaging ofσ1receptor in the brain will enable the elucidation of this receptor chaperone’s involvement in neurologic and psychiatric disorders and its changes during disease progression, as well as exploration of therapeutic mechanism to facilitate new drug development.
Currently, there is noσ1receptor radiotracer approved for use in clinical routine.[11C]SA4503 is the first radioligand used for imagingσ1receptor changes in the brain of AD [27] and PD[28] patients andσ1receptor occupancy by fluvoxamine [29] and donepezil [30].But the short half-life of its11C-nuclide limits its applications.Among the18F-labeled radioligands investigated in humans, [18F]FPS [31] and [18F]FTC-146 [32,33] exhibit irreversible kinetics, which excludes their applications for the imaging and quantification ofσ1receptors in the human brain.Currently, only(S)-[18F]fluspidine [34] has proved to be a promising radioligand and used for imagingσ1receptor in patients with major depression [35] andσ1receptor occupancy of pridopidine [36].Nonetheless, its free fraction in plasma is low (2.3%), making reliable measurement challenging, and further validation is required to test its reproducibility in quantifying theσ1receptor in humans.Therefore, there is a need for the development ofσ1receptor radiotracer with optimal pharmacokinetic and imaging properties.
We have recently reported the characterization of [18F]FBFP([18F]1) in non-human primates that demonstrated high plasma fraction, good brain uptake, and the highest specific binding signals in non-human primates among theσ1receptor radioligands evaluated to date [37,38].Similar to [18F]fluspidine, [18F]1 has a chiral center and thus is composed of two enantiomers as shown in Fig.1.For future clinical translation a single enantiomer is desirable, as the two enantiomers may possess different affinity and hence pharmacokinetic and imaging characteristics, as in the case of (R)- and (S)-[18F]fluspidine [39–41].Therefore, we synthesized the two enantiomers of FBFP (1) forin vitrobinding assays and agonist/antagonistic activity test.The18F-labeled enantiomers (S)-and (R)-[18F]1 were also synthesized and evaluatedviabiodistribution in mice and micro-PET imaging studies in rats.
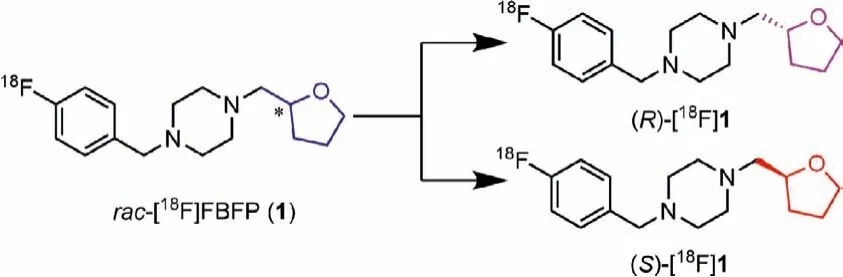
Fig.1.Structures of 18F-labeled σ1 receptor enantiomers (S)- and (R)-[18F]1.
The synthetic routes for the two enantiomers (S)-1 and (R)-1 and enantiopure precursors for (S)- and (R)-[18F]1 were illustrated in Schemes 1 and 2.The racemic compoundrac-1 andrac-5 were prepared following our previously reported methods [37] with minor modifications, resulting in a shorter synthesis time and higher yield.Separation ofrac-1viachiral preparative HPLC provided the two enantiomers (S)-1 and (R)-1.The detailed chiral HPLC conditions for separation are presented in Table S1 (Supporting information).
To obtain the single enantiomer (S)-1 or (R)-1 directly,we used (S)-(+)- or (R)-(-)-tetrahydrofurfuryl alcohol ((S)-6)or (R)-6) as starting material, which was converted toptoluenesulfonate (S)-7 or (R)-7 for nucleophilic substitution with LiBr to give the bromo–intermediate (S)-8 or (R)-8.N-Alkylation of piperazine (9) with 4-halide-benzyl bromide (3 or 4) provided the intermediate 10 or 11.FurtherN-alkylation with (S)-8 or (R)-8 then afforded (S)-1, (R)-1, (S)-5 or (R)-5, respectively.
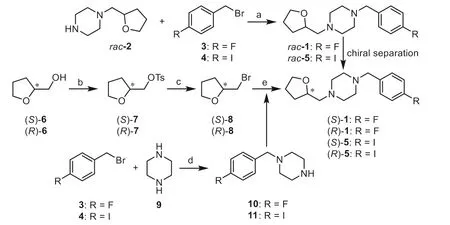
Scheme 1.Synthetic routes for rac-1 and its two enantiomers (S)- and (R)-1.Reagents and conditions: (a) K2CO3, KI, CH3CN, 40 °C, 2 h, 77%-91%; (b) TsCl, TEA,DMAP, CH2Cl2, 0 °C to r.t., overnight, 94%-99%; (c) LiBr, acetone, reflux, 24 h, 56%;(d) TEA, CH3OH, 60 °C, 3 h, 72%-80%; (e) 10 or 11, K2CO3, NaI, DMF, reflux, 2 h,76%-91%.
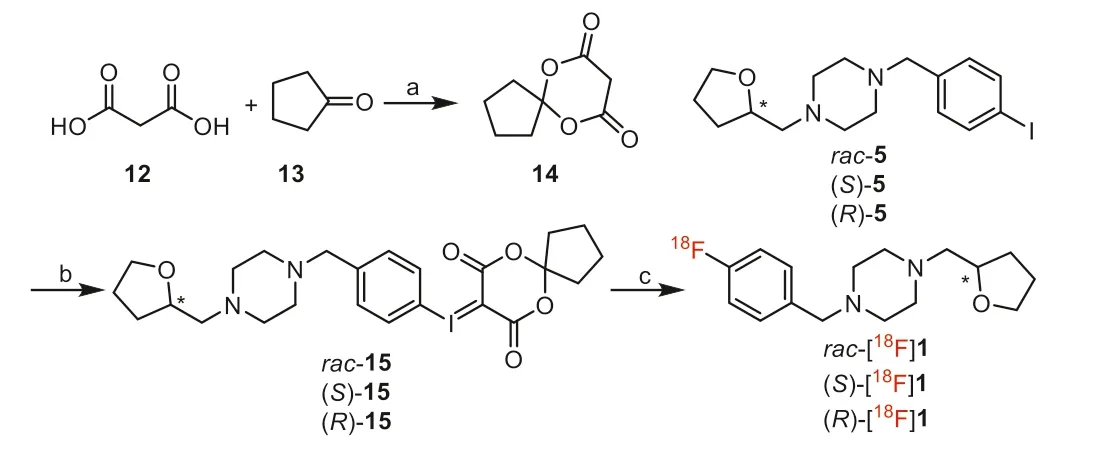
Scheme 2.Synthesis of the iodonium ylide precursors and radiosynthesis of (S)-and (R)-[18F]1.Reagents and conditions: (a) Ac2O, H3BO3, 30 °C, 0.5 h, 32%; (b) i)TFA, CHCl3, Oxone, r.t., 1 h; ii) 6,10-dioxaspiro[4.5]decane-7,9–dione (14), Na2CO3(10% aq), EtOH, 35 °C, 0.5 h, 77%; (c) [18F]F-, Kryptofix 2.2.2 (K2.2.2), K2CO3, TPP,DMF, 120 °C, 10 min.
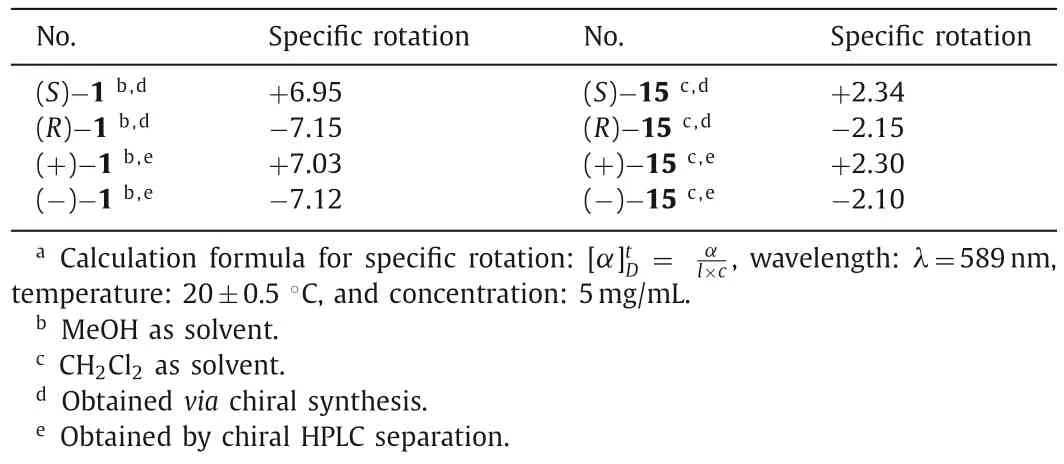
Table 1 Specific rotation and absolute configuration of the enantiomers.a
The iodonium ylide precursorsrac-15, (S)-15 and (R)-15 were synthesized based on our previously reported methods [38] with minor modifications as shown in Scheme 2.Using boric acid(H3BO3) as catalyst [42] instead of concentrated sulfuric acid [43],6,10-dioxaspiro[4.5]decane-7,9–dione (14) was obtained in a simple and efficient method.Similar to the synthesis of the two enantiomers of compound 1, (S)-15 and (R)-15 were obtained by separation ofrac-15viachiral preparative HPLC (Table S2 and Fig.S2 in Supporting information), orviachiral synthesis from (S)-5 or(R)-5.
The final products were analyzed by chiral HPLC and determined to have more than 98% enantiomeric excess (ee) for (S)-1,(R)-1, (S)-15 and (R)-15, as shown in Figs.S1 and S2 (Supporting information).
The optical rotations of the enantiomers (S)-1 and (R)-1 and radiolabeling precursors (S)-15 and (R)-15 was determined by a polarimeter and shown in Table 1.The specific rotationswere +6.95 and -7.15, respectively, for (S)-1 and (R)-1, prepared by chiral synthesis.Values for the samples (+)-1 and (-)-1, obtained by chiral HPLC separation, were similar (+7.03 and -7.12,respectively).Thevalues for (S)-15 and (R)-15 (+2.34 and-2.15, respectively) from chiral synthesis were also similar to those of (+)-15 and (-)-15 (+2.30 and -2.10, respectively), obtained by chiral HPLC separation.
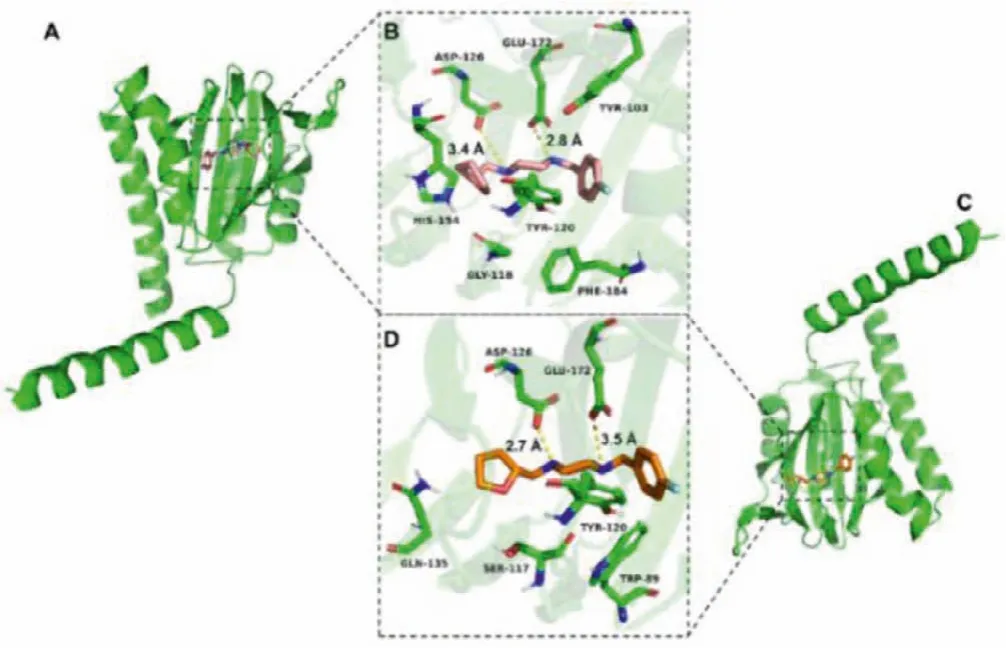
Fig.2.The docked poses of the enantiomers (S)-1 and (R)-1 into σ1 receptor.(A) and (B) Pose of co-crystallized (S)-1 and top-ranked docking (Carbon: pink;Nitrogen: blue; Oxygen: red; Fluorine: pale cyan).(C) and (D) Best docked pose for(R)-1 (Carbon: orange; Nitrogen: blue; Oxygen: red; Fluorine: pale cyan).Hydrogen bonds are indicated by yellow dashed lines.In all panels, the σ1 receptor is shown in green.
The structure of the humanσ1receptor has been reported [3].Molecular docking studies were conducted to investigate and predict the possible molecular interaction mode of the enantiomers(S)- and (R)-1 with the humanσ1receptor.The method for docking models was referenced and revised from previousσ1receptor ligand recognition studies [3,5,44].
It is well known that the electrostatic and steric-hindrance interaction between the basic nitrogen atom on the ligand scaffold and Glu-172 or Asp-126 are significantly important.Both enantiomers (S)- and (R)-1 were determined to be suitable for the binding pocket ofσ1receptor occupied with a similar pose (Figs.2A and C).As depicted in Figs.2B and D, both enantiomers featured hydrogen-bond interactions between the two nitrogen atoms of the piperazine ring with Glu-172 and Asp-126.However, the distances between the nitrogen atoms and the two amino acid residues above are different.The distances between the nitrogen atom of the piperazine adjacent to the 4-fluorobenzyl group and Glu-172 are 2.8and 3.5for (S)-1 and (R)-1, respectively.The distances between the nitrogen atom adjacent to the tetrahydrofurfuryl group and Asp-126 are 3.4and 2.7for (S)-1 and (R)-1,respectively.Such notable differences for the two enantiomers could affect their pharmacokinetics and dynamicsin vivo.The values of the receptor-ligand scoring function were approximately-10.00 and -9.88 kcal/mol for the enantiomers (S)-1 and (R)-1,respectively.The two-dimensional diagram of receptor-ligand binding is presented in Fig.S3 (Supporting information).
Competition binding assays were performed as previously reported [45], using rat brain homogenates with (+)-[3H]pentazocine as radioligand for theσ1receptors and rat liver membranes with[3H]DTG (in the presence of 10 μmol/L dextrallorphan to blockσ1receptors) as radioligand for theσ2receptors, respectively.The results are shown in Table 2.TheKi(σ1) values were 3.22±0.87,3.16±1.07 and 3.23±0.62 nmol/L, respectively, forrac-1, (S)-(+)-1 and (R)-(-)-1.TheKi(σ2) values were 168±56.0, 126±5.13 and 178±32.8 nmol/L, respectively, forrac-1, (S)-(+)-1 and (R)-(-)-1.TheKi(σ2)/Ki(σ1) ratios were 52, 40 and 55, respectively, forrac-1, (S)-(+)-1 and (R)-(-)-1.These data indicated that both enantiomers possessed comparable low nanomolar affinity forσ1receptors and high subtype selectivity.

Table 2 Binding affinities of rac-, (S)- and (R)-1 for σ1 and σ2 receptors.a
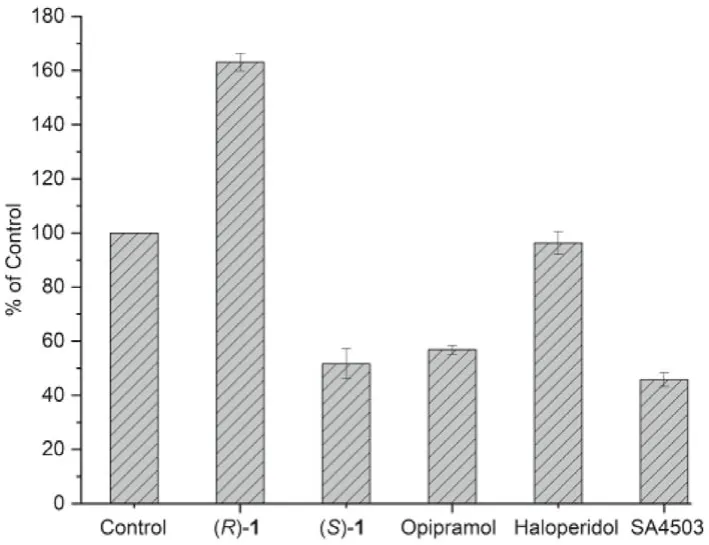
Fig.3.Effect of ligands (S)-1, (R)-1 and σ1 receptor agonist opipramol, SA4503 and antagonist haloperidol on KCl-induced Ca2+ influx.For the control group, the PC12 cells were incubated with 0.5% DMSO (solvent used to dissolve the compounds for this test) for 10 min, and then stimulated with KCl (80 mmol/L) for 10 min.
Agonists and antagonists of theσ1receptor have been shown to bind with different oligomeric states, and hold different therapeutic potential in clinical trials [5,46].Agonists stabilizeσ1receptor monomers and dimers that are the active forms of the chaperone protein, whereas antagonists bind to higher oligomer complexes [6,7].Based on the report thatσ1agonists can inhibit potassium chloride (KCl)-induced Ca2+influx into synaptosomes, an assay to determine the agonistic/antagonistic effects ofσ1ligands has been established using opipramol, aσ1agonist which can reduce KCl-induced Ca2+influx, by fluorescence measurements with fura-2 or fura-4-AM [47–49].Given the inhibition of Ca2+influx by agonists, we use fura-4-AM imaging to measure calcium response and test agonist/antagonist activity for (S)- and (R)-1.The results are shown in Fig.3.Compared to control, theσ1receptor agonist opipramol and SA4503 inhibited Ca2+influx by 43% and 54% (P <0.001), respectively.Similar to opipramol and SA4503, (S)-1 was able to reduce the KCl-induced Ca2+influx by 48% (P <0.001),indicating (S)-1 asσ1receptor agonist.However, theσ1receptor antagonist haloperidol had no inhibition effect on ion reflux(P=0.204).(R)-1 further increased the KCl-induced Ca2+influx by 63% (P <0.001).
In the second experiment, synaptosomes were preincubated with the test compound (R)-1, (S)-1 and SA4503 for 5 min.Then, opipramol was added and after 5 min, the cells were stimulated with KCl.The effect of ligands (S)-1, (R)-1 andσ1receptor agonist SA4503 on opipramol-induced inhibition of Ca2+influx is shown in Fig.S4 and Table S3 (Supporting information).Preincubation with theσ1receptor agonist SA4503 strengthened opipramol inhibition of Ca2+influx (31% increase compared to opipramol alone,P=0.002).Preincubation of (S)-1 had no significant effect on opipramol inhibition of Ca2+influx (21% decrease,P=0.295).It is notable that preincubation with (R)-1 reversed the effect of opipramol on Ca2+influx significantly (218% compared to opipramol group,P <0.001), indicating (R)-1 as aσ1receptor antagonist.Both experiments together demonstrate theσ1antagonistic effect of (R)-1 and theσ1agonistic effect of (S)-1.Differentσ1agonist/antagonist activity for (S)- and (R)-1 warrant further investigation for their therapeutic applications.
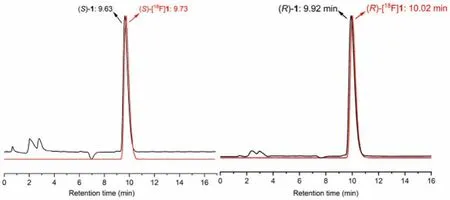
Fig.4.HPLC profiles from co-injection of (R)-1 (tR=9.92 min) with (R)-[18F]1(tR=10.02 min), and (S)-1 (tR=9.63 min) with (S)-[18F]1 (tR=9.73 min).HPLC analyses were performed on the Shimadzu SCL-20 AVP system.Conditions: Agela Venusil MP C18 column (250×4.6 mm, 5 μm), 50% acetonitrile and 50% water containing 0.05% triethylamine (TEA) at 1 mL/min.
One-step radiosynthesis of racemic and two enantiomers of[18F]1 were explored based on the previously reported method[38] with different conditions as shown in Scheme 2.Chiral HPLC analysis indicated no racemization during the radiosynthesis.The two enantiomers (S)- or (R)-[18F]1 were obtained in more than 98% enantiomeric excess (ee) from the corresponding enantiopure iodonium ylide precursors ((S)-(+)-15 and (R)-(-)-15), as shown in Fig.S5 (Supporting information).Moreover, with triphenylphosphine (PPh3, TPP) as a ligand/catalyst, the radiochemical yields(RCY) were significantly improved (decay-corrected RCY of 24.4% ±2.6%,n=6,vs.10% as previously reported [38]) with lower reaction temperature (120 °Cvs.150 °C) and shorter reaction time (10 minvs.25 min).Rac-, (S)- and (R)-[18F]1 were obtained with>99% radiochemical purity (RCP) and molar activity of 86–214 GBq/μmol.
To identify the radiotracers, the corresponding unlabeled compounds were co-injected and co-eluted.The HPLC profiles of (S)-and (R)-[18F]1 are presented in Fig.4.The retention times of (R)-1 and (R)-[18F]1 were 9.92 and 10.02 min, respectively.The retention times of (S)-1 and (S)-[18F]1 were 9.63 and 9.73 min, respectively.The difference in retention times was in accordance with the time lag as a result of the volume and flow rate within the distance between the UV and radioactivity detectors of our HPLC system.
Biodistribution studies with (S)- and (R)-[18F]1 (185–296 kBq,0.1 mL, 7% ethanol in saline) were performed in male ICR mice to test if there are differences in kinetics.The results are illustrated in Tables S4 and S5 (Supporting information).Similar torac-[18F]1,both (S)- and (R)-[18F]1 exhibited high initial uptake in the brain(>10% ID/g at 2 min) and low levels of blood accumulation after 30 min.Thus, both enantiomers possessed high brain-to-blood ratios of around 20 from 15 min to 60 min.Radioactivity accumulation in the bone was low at 60 min (<5% ID/g) and remained constant with time, indicating no defluorination of (S)- and (R)-[18F]1in vivo.
To determine the binding specificity of (S)- and (R)-[18F]1in vivo, blocking studies with theσ1selective agonist SA4503 and self-inhibition by (S)- or (R)-1 were conducted.SA4503 (0.1 mL,5 μmol/kg) and unlabeled (S)- or (R)-1 (0.1 mL, 5 μmol/kg) were injected 5 min prior to radiotracer injection.The results are summarized in Fig.5.Similar torac-[18F]1, pretreatment with SA4503 led to a significant reduction in brain uptake of both (S)-[18F]1(83%,P <0.001) and (R)-[18F]1 (78%,P <0.001).Meanwhile, radioactivity accumulation in the blood were significantly increased by 195% and 231%, respectively, leading to decreased brain-toblood ratios (94% reduction for both enantiomers,P <0.001).Moreover, pretreatment with SA4503 also resulted in significant reduction of radiotracer uptake in peripheral organs known to containσ1receptors, including the heart (69% and 67% reduction for(S)- and (R)-[18F]1,P <0.001), spleen (71% and 62%,P <0.001),lung (83% and 76%,P <0.001), pancreas (47% and 50%,P <0.001)and muscle (57% and 38%,P <0.001).For self-blocking studies,pretreatment with unlabeled (S)-1 or (R)-1 at the same dose of SA4503 led to significant reduction in brain uptake (88% and 82%,P <0.001), and brain-to-blood ratios (96% and 95%,P <0.001),as well as significant reduction of uptake in peripheral organs including the heart (72% and 70%,P <0.001), spleen (76% and 60%,P <0.001), lung (86% and 74%,P <0.001), pancreas (31% and 39%,P <0.001) and muscle (62% and 49%,P <0.001).These results demonstrate high levels of specific binding for both (S)-[18F]1 and(R)-[18F]1 toσ1receptorsin vivo.
The metabolic stability of (S)- and (R)-[18F]1 was investigatedin vivoin male ICR mice at 30 min post-injection of the radiotracers(74–92.5 MBq, 0.1 mL, 7% ethanol in saline).Representative HPLC chromatograms of the acetonitrile extracts obtained from plasma and brain samples are presented in Fig.6.In the brain samples,>95% of the radioactivity signal represented the intact parent radiotracer for both (S)- and (R)-[18F]1 (n=3).Only small amount of a hydrophilic radioactive metabolite was observed with retention times of 2.63 and 2.84 min for (S)- or (R)-[18F]1 (Figs.6B and E), respectively.In plasma samples, only one major hydrophobic radio-metabolite withtRof 2.61 and 2.88 min, respectively, were detected after injection of (S)- or (R)-[18F]1 (Figs.6C and F).
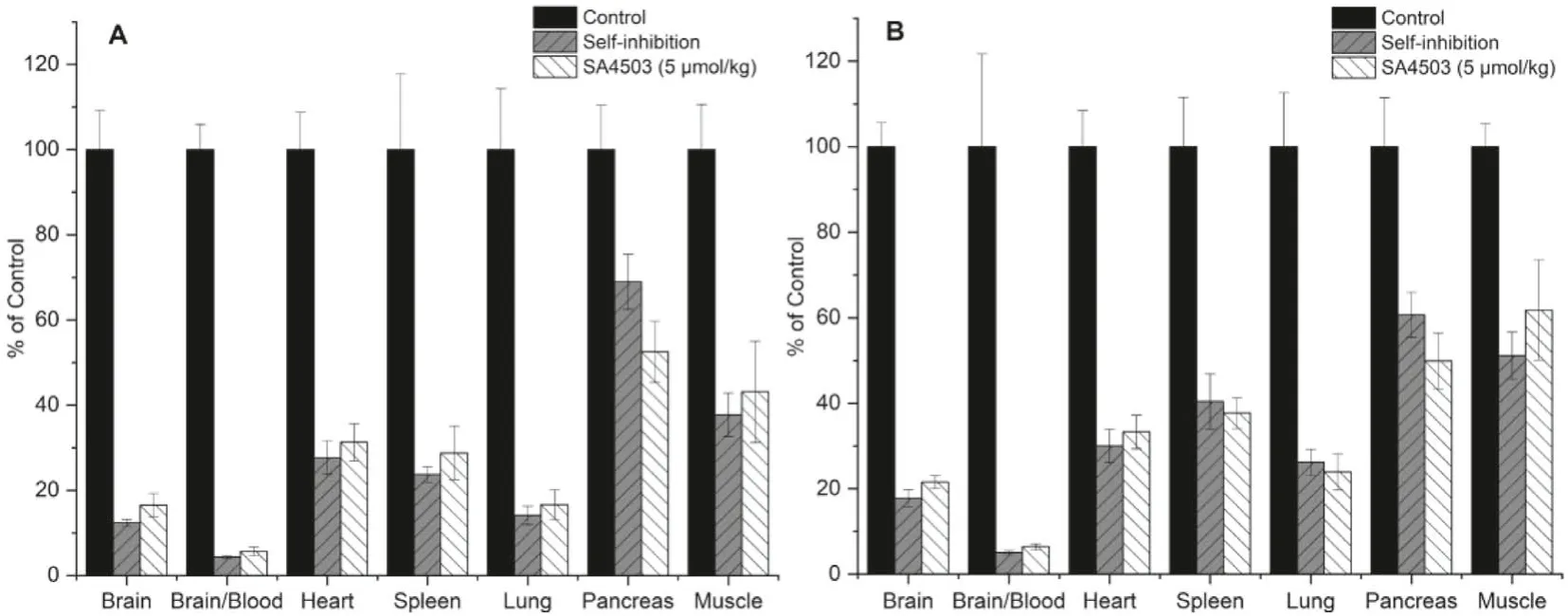
Fig.5.Effects of pretreatment with (S)-1 (0.1 mL, 5 μmol/kg), (R)-1 (0.1 mL, 5 μmol/kg) and SA4503 (0.1 mL, 5 μmol/kg) on organ biodistribution of (S)-[18F]1 (A) and(R)-[18F]1 (B) at 30 min after intravenous injection.Values are means ± SD, n=8.
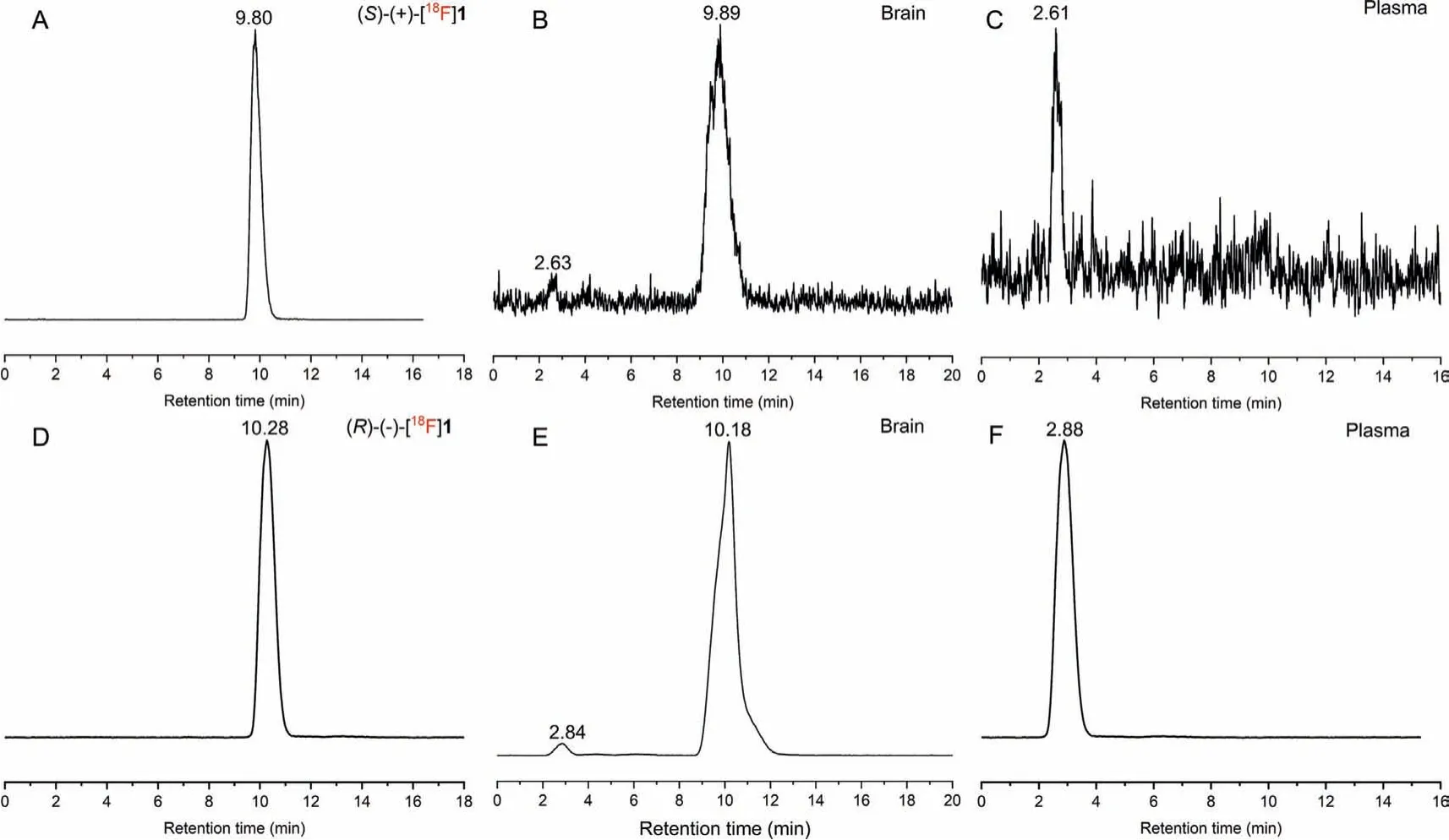
Fig.6.Analytical radio-HPLC chromatograms of the mouse plasma and brain extracts at 30 min after administration of (S)- [18F]1 (A: (S)-[18F]1; B: brain extracts; C: plasma extracts) and (R)-[18F]1 (D: (R)-[18F]1; E: brain extracts; F: plasma extracts).
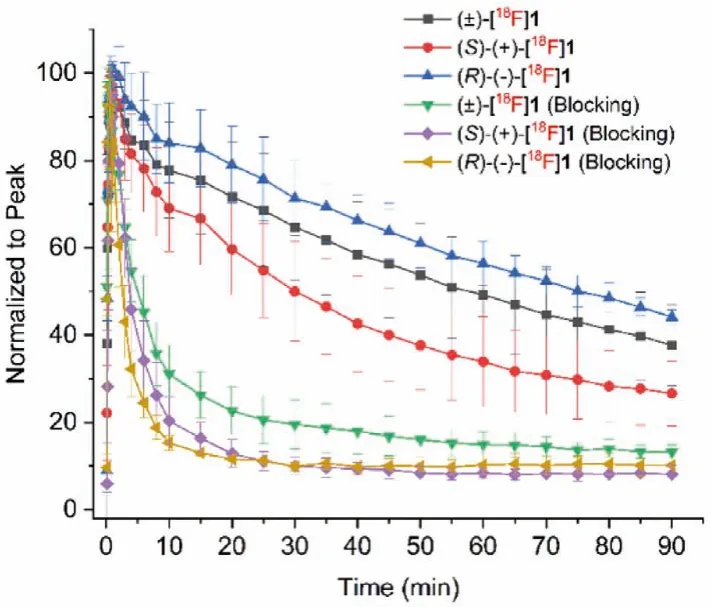
Fig.7.Time–activity curves of rac-, (S)- and (R)-[18F]1 in the whole brain from baseline (n=3–7) and SA4503 blocking scans (n=3) in Sprague-Dawley rats.
To further investigate the kinetics and confirm the specific binding of two enantiomers, micro-PET/CT studies were conducted in anaesthetized Sprague-Dawley (SD) rats.In vivobinding specificity was evaluatedviaco-injection of SA4503 (5 μmol/kg) with the radiotracers (7.40–9.25 MBq, 0.5 mL, 7% ethanol in saline).Dynamic brain PET scanning was started immediately after injection of the radiotracers and lasted for 90 min.The time-activity curves(TACs) ofrac-, (S)- and (R)-[18F]1 in the whole brain from baseline (n=6, 7, 3, respectively) and SA4503 blocking scans (n=4, 3,3, respectively) are presented in Fig.7.Similar torac-[18F]1, (S)-and (R)-[18F]1 entered the brain rapidly and reached peak concentrations within 2 min and washed out steadily over time.However,(S)-[18F]1 exhibited much faster clearance than (R)-[18F]1 from the rat brain.The different kinetics of (S)-(+)- and (R)-(-)-[18F]1 in the brain of mice and rats may be related to species difference.Pretreatment with SA4503 resulted in significant reduction of radiotracer uptake, confirming high specific binding of both enantiomers toσ1receptors in the rat brain.
In conclusion, we have developed efficient procedures for the synthesis of enantiomerically pure ligands (S)-1 and (R)-1 and the corresponding iodonium ylide precursors.Computational docking studies indicated both enantiomers as suitable ligands for theσ1receptors.Moreover, the molecular interactions between (S)-1 and theσ1receptor was found to be different from that between (R)-1 and theσ1receptor.It is also interesting that (R)-1 behaved as an antagonist while (S)-1 as an agonist.The enantiomerically pure radioligands (S)-(+)- and (R)-(-)-[18F]1 were obtained from their corresponding iodonium ylide precursors with improved radiochemical yields using triphenylphosphine as a ligand/catalyst.Evaluation in rodents demonstrated excellent properties of both (S)-(+)-[18F]1 and (R)-(-)-[18F]1 with high brain uptake, high brainto-blood ratios, high metabolic stability in the brain and high specific binding to theσ1receptors.In dynamic micro-PET studies,(S)-(+)-[18F]1 exhibited much faster clearance from the rat brain compared to (R)-(+)-[18F]1, demonstrating different pharmacokinetics of two enantiomerically pure radioligands.Taken together,(S)-(+)-[18F]1 and (R)-(-)-[18F]1 warrant further evaluation in primates to develop an optimal enantiomerically pure radioligand for imaging ofσ1receptors in humans.
Ethics statement
All procedures related to the animal experiments were performed in compliance with relevant laws and institutional guidelines.All the animal experiments of the ICR mice and rats were approved by the Institutional Animal Care and Use Committee of Beijing Normal University.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the financial support from Beijing Natural Science Foundation (No.7212203) and National Natural Science Foundation of China (No.21876013).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.099.
 Chinese Chemical Letters2022年7期
Chinese Chemical Letters2022年7期
- Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
- Recent development in selective Tau tracers for PET imaging in the brain
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging