
Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
2022-07-09 02:14FeizeLiYuanyouYangJialiLiaoNingLiu
Chinese Chemical Letters 2022年7期
Feize Li, Yuanyou Yang, Jiali Liao, Ning Liu
Key Laboratory of Radiation Physics and Technology of the Ministry of Education, Institute of Nuclear Science and Technology, Sichuan University, Chengdu 610064, China
ABSTRACT Astatine-211 (211At, t1/2=7.21 h) emitting two α particles with energies of 5.87 and 7.45 MeV, can lead to a high linear energy transfer (LET=98.84 keV/μm) and short tissue range (50~90 μm).Since the 1950s,211At had stepped into endoradiotherapy and has always been regarded as one of the most promising αemitters for targeted-alpha therapy (TAT) in various malignancies.In the past two decades, 211At related radiopharmaceuticals have achieved great progress in the studies of basic physicochemical properties of astatine, 211At labeling strategies, preclinical and clinical studies, producing profound effects in nuclear medicine.This work will give a panorama of 211At-related researches in the recent 20 years, which will cover both the fundamental insights of 211At radiochemistry and applied labeling compounds.It can provide some important hints for the studies of TAT and other radiopharmaceuticals applied in tumor radiotherapy.
Keywords:Radionuclides Astatine-211 Target-alpha therapy Radiopharmaceuticals
1.Introduction
Recently, targeted-alpha therapy (TAT) has drawn much attention both in preclinical and clinical researches.The LET ofαparticles is ~100 keV/μm, which is about 500 times higher than that of widely-usedβ-particles.High LET value enablesαparticles to more effectively trigger DNA double-strand breaks (DSBs), thus inducing cell cycle arrest and leading to mitotic cell death, apoptosis or necrosis [1,2].Meanwhile, the path length ofαparticles(50 ~100 μm) is equivalent to the distance of 5 ~10 cells, which can reduce the irradiation to surrounding healthy cells and unnecessary damage to normal tissues and organs [3].These excellent properties make TAT show great advantages in the treatment of minimal residual tumors after primary treatment including surgery, external radiotherapy and chemotherapy.
There are over 2000 known radionuclides, and about 400 areαemitting.Idealα-emitters for TAT should have several basic characteristics [4]: (1) suitable half-lives for radiolabeling, purification and transportation, (2) contain no long-lived toxic decay daughters, (3) and there are available production methods for clinical applications.These criteria will screen all candidates to no more than ten, including149Tb,211At,212/213Bi,223/224Ra,225Ac,226/227Th and230U (Table 1) [5,6].Among all these availableαnuclides,211At has unique advantages in terms of nuclear properties (Fig.1).The half-life of211At (t1/2=7.21 h) is long enough for multi-step radiolabeling and is also compatible with plenty of carriers, such as small molecules, peptides and antibody/fragments [7].211At is regarded as a 100%α-emitter with twoαparticles with energies of 5.87 and 7.45 MeV.Despite its potential uptake by bone, liver and kidney, the toxicity of207Bi can be negligible [8]: 370 MBq of211At, the currently highest dose administered to a human, only leads to about 3.7 kBq of207Bi.There is a chemical conversion of astatine to polonium which will cause the radionuclide to detach from the carriers, but the211Po daughter is not affected by nuclear recoil since it comes from211At by electron capture (EC).Even it can immediately escape from the cell surface and diffuse freely,211Po with a short half-life of 0.52 s tend to decay to207Pb within two cell diameters beyond the original cell surface [9].In contrast, when211At decays into211Po by EC, the characteristic Xrays of 77~92 keV can be used for measuring211At byγ-detectors in laboratory or clinical centers, which are also available for imaging by planar or SPECT scanners [10–13].Additionally,211At can be produced by bombarding natural Bi withαparticles (≥20 MeV)on a cyclotron, which makes it more available than most medicalα-emitters.
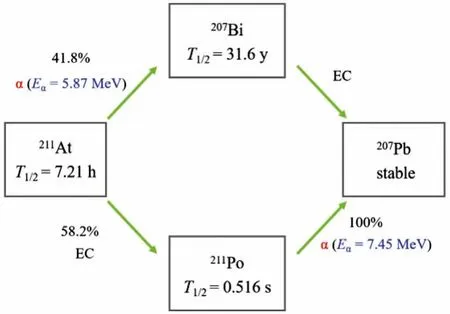
Fig.1.Decay diagram of 211At.
Actually, in 1950, Hamiltonet al.[14] found that211At could induce extreme degree of radiation injury of the thyroid gland in the rat without apparent involvement of the adjacent parathyroid gland, demonstrating that thisα-emitter might be of essen-tial therapeutic application in hyperthyroidism.Since then,211At had stepped into endoradiotherapy and kept drawing more and more attention in nuclear medicine.Especially,211At-related radiopharmaceuticals have made great progress since 2000.It has been demonstrated that211At is one of the most promisingαnuclides for TAT and corresponding radiopharmaceuticals have widely-clinical prospects.Although several breakthroughs about211At-labelled pharmaceuticals have been involved in some of the latest comprehensive reviews focused on introducing TAT or endoradiotherapy [15,16], there is still a lack of specific work illustrating the new developments about211At-based radiotherapy over the past decade, as the latest one can be dated to 2013 [8].Considering newly emerging advances in211At-based medicines should have profound effect in TAT, especially those involving the fundamental properties and clinical trials, we believe that it is very important to retrospect recent progresses of211At again.
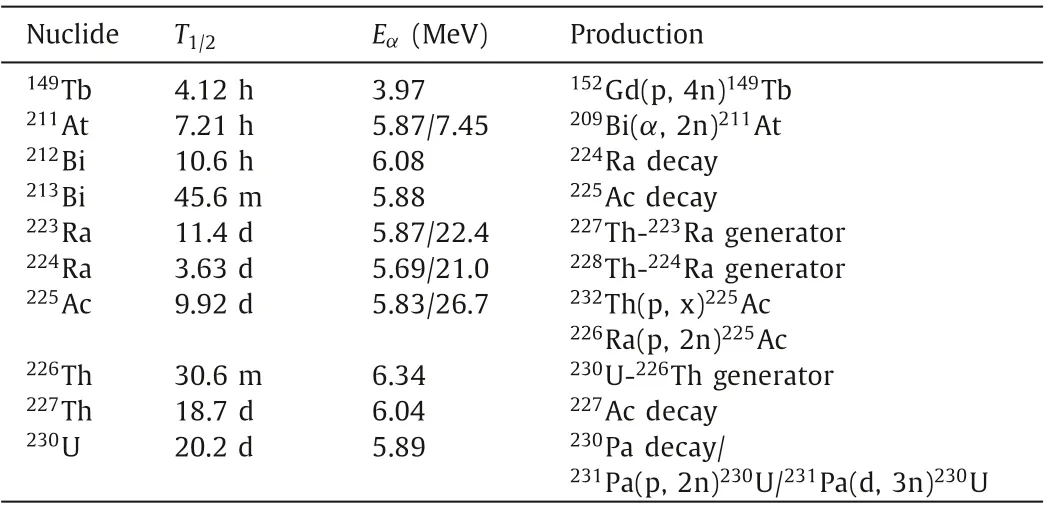
Table 1 Several α-emitters suitable for TAT [5,6].
In this review, we will introduce the progress of211At about the basic properties of astatine, the separation methods of211At, the brief strategies for211At labeling and the preclinical and clinical trials about related targeted pharmaceuticals.It will mainly cover most of the work from 2000, since ~70% of the publications involving211At are located in 2000~2021, when astatine/astatine-211/At-211/211At is typed as the topic to search articles throughweb of sciencewithout other limitations (on February 28, 2022).In addition, despite of considerable amount of the work involving211At in 1940~2000 focused on the nuclear properties of astatine,there is already a review from Zalutskyet al.[7] in 2000 specializing on introducing211At-labelled radiopharmaceuticals.The goals of this work include: (1) providing some general introduction of211At to the newcomers in TAT, so that they can recognize both the prospects and challenges of211At-related radiopharmaceuticals;(2) systematically concluding the progress of211At-based radiotherapy in recent years for researchers who are engaging in radiochemistry and nuclear medicine; (3) summarizing some prob-lems before clinical translation of211At-labelled compounds, and comprehensively evaluate the application potential of211At.We believe that this work is not only helpful to the development of211At-labelled radiopharmaceuticals, but can also provide some important hints for endoradiotherapy researches based on otherαemitters.
2.Basic properties of astatine
2.1.Isotopes of astatine
Astatine (Z=85), located at the seventh group of the periodic table, was discovered in 1940 by Corsonet al.[17].It was obtained by bombarding natural Bi target with 32 MeV ofαparticles and was initially named as “eka-iodine”, due to its high similarity to iodine.In 1947, it was formally identified as astatine, from the ancient Greek “astatos” which means instability [18].Astatine is often considered one of the rarest of all elements, and the total amount of natural astatine at any given time range is estimated from a few hundred milligrams to 30 g.Among all isotopes of astatine,214–219At occurs naturally in the decay chains of transuranium elements, and the most abundant one is217At (t1/2=32.6 ms) decayed from237Np [19].Other isotopes of astatine are artificially synthesized, and210At has the longest half-life of 8.1 h.The isotopes of astatine with the mass lighter than 217 decay by pureα-emission or by a mixture ofα- and EC/β+-emissions,217–220At undergo bothα- andβ--emissions, while astatine isotopes with mass>220 decays by pureβ--emission (Table 2).
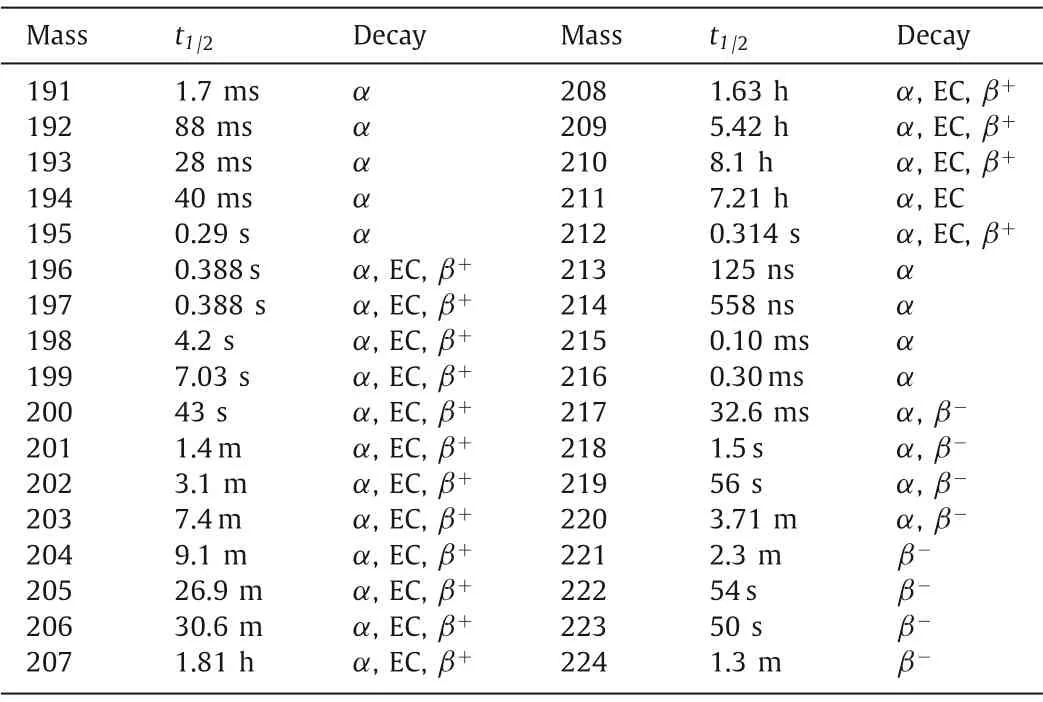
Table 2 The nuclear properties of 191–224At.
Although207–210At with suitable half-lives (>1 h) can emitα,β+as well asγrays, therefore show possible application in nuclear medicine, the prospect of these radionuclides is severely clouded by some other factors.For207/208At, there is no suitable way for producing medically-required amount of such radionuclides.It is previously-reported that209At might be suitable for SPECT imaging in nude mice [20], but the production of209At requires high-energy protons (480 MeV) to bombard uranium carbide targets [21].The production of210At is not an serious issue and it was ever used to replace211At asγ-emitter to quantify thein vivodistribution of astatine [22], but its daughter210Po (t1/2=138.38 d) is extremely toxic, making this nuclide almost have no prospect in biomedicine.Similarly, theα-emitters208/209Po (t1/2=2.90 and 124 y) in the decay chains of208/209At also limit their medical practice: once entering the organism, they may also cause long-term unnecessary internal irradiation and corresponding biotoxicity.As a result, only211At has currently been employed in radiopharmaceuticals and been receiving more and more attention.
2.2.Physicochemical properties of At
As pointed out by Kozimoret al.[23], At should be one of the least understood elements in the periodic table.The most important reason is that, even for211At with relatively long half-life and nontoxic decay daughters, it can be safely produced and operated with the amount limited to be 37 kBq to 4 GBq, roughly equivalent to 4.8×10-13g to 5.2×10-8g (rarely reaches the upper limit).Such quantities are beyond the detection limits of the most sensitive mass spectrometry, not to mention NMR, EPR, UV–vis and X-ray spectroscopy.Besides, for most chemical reactions,211At currently ranges from 10-13g to 10-9g, and may even be smaller than the amount of trace organic species and metals in solvents.Thus,these intrinsic trace impurities may interfere during related reaction even catalyze unexpected reaction pathways.
According to periodic law, it seems feasible to use I as an analogue of At in fundamental study.However, this strategy must be cautiously taken since At is semi-metallic [24–26], while I is absolutely non-metallic.There are also theoretical studies showing that Al13cluster and At possess similar chemical reactivity [27], especially in electron transfer process.Meanwhile, both of them can form stable compounds with isobutylbenzene.Obviously, this kind of analogy at atomic levels is difficult to be extrapolated to macroscopic system.For the most widely-used211At, 1 MBq equals just 0.06 pmol atoms, making it statistically unlikely to exist as211At2.Up to now, the color of astatine is not even known and is speculated to be black based on the fact that halogens from fluorine to iodine get darker [28].The melting and boiling points of At are estimated as 302 and 337 °C, respectively [4].On silica, the adsorption enthalpies of At, AtO2and HAtO determined by Serovet al.[29] are 124 ± 5, 80 ± 5 and 47 ± 5 kJ/mol, while the sublimation enthalpies are 148 ± 25, 106 ± 30 and 56 ± 30 kJ/mol, respectively.On the gold surface, the adsorption enthalpies of At and AtO2were determined to be 147 ± 15 and 124 ± 5 kJ/mol, respectively.
The behavior of At in different solutions is also complicated.211At is stable in chloroform and loses no activity even when the solvent is completely evaporated.However, the species distribution in chloroform is very variable.At can react with chlorine and other oxygen-containing radiolysis products derived from chloroform, producing a variety of species in a dose-dependent manner [30].It has been demonstrated that the volatilization of free211At is an heterogeneous process and 2.3 times higher than that of Na211At even in confined space [31].This may be tightly related to the various oxidation states of At (-1, +1, +3, +5, +7) [32], which also lead to multiple species in aqueous solution,e.g., At-, At(0),AtO-, AtO+, AtO2-, AtO3-and AtO4-].However, for a long time,the specific species of At lacked relevant experimental data to support.Recently, Guoet al.[33] successfully measured the absolute movement velocity of At monovalent anion in acidic reducing condition to be -8.26 ± 0.59×10-4cm2V-1S-1, which was highly coherent with theoretical calculation and directly proved the existence of At-.Championet al.[34] confirmed that At(0) cannot exist in aqueous solution, while At(-I), At(+I) and At(+III) should belong to At-, At+and AtO+in the range of pH 1~2, with the standard redox potentials of At+/At-and AtO+/At+pairs being 0.36 and 0.74 (0.01 Vvs.NHE), respectively.It has also been demonstrated that the main species of At at pH 11 is AtO(OH)2-but not At-[35].Meanwhile, AtO+is able to form a neutral substance AtO(OH)mainly through the reaction of AtO++H2O(l) ⇌AtO(OH)(aq)+H+when increasing solution pH, and the equilibrium constant was 10-1.9[36].At+can form stable ternary complexes IAtBr-in aqueous solution, with an equilibrium constant of 107.5±0.2[37].Nishinakaet al.[38] speculated that At can also exist as AtO3-and AtO4-in the presence of strong oxidizer (such as KIO4).However, this result was based on the comparison ofRfvalues between At and I species (IO3-and IO4-) using thin layer chromatography(TLC) methods, which still needs more direct evidences.
As a typical intermolecular interaction, halogen bond is formed by attraction between a halogen atom X from a molecule or a molecular segment R-X (R is usually more electronegative than X or is X itself) and another atom from R-X or a different molecule B.In a typical case, B is usually a Lewis base, and R can be another halogen atom or an inorganic/organic residue [39].Loosely,halogen bonds occur when there are attractive interaction between an electrophilic region associated with a halogen atom in one molecular entity and a nucleophile region in another or the same molecular entity [40].Guoet al.[41,42] demonstrated that AtI can interact with various Lewis bases through halogen bond,and it is a stronger halogen bond donor than I2.They also found that tributylphosphine oxide (Bu3PO) and AtI can form either 1:1 or 2:1 complex [43].The former has the currently strongest halogen bond, and the formation constant is as high as 104.24±0.35.For the 2:1 complex, a Bu3PO molecule is first linked to AtI by halogen bond, while another one participated in the formation of the terpolymer by forming hydrogen bond with the organic entity.
The metallic property of At has encouraged researchers to look for suitable chelating agents which can form stable At-centered coordination complexes.A study performed by Bassalet al.[44] involving more than 20 model ligands indicated that those contain S and O donors exhibit higher affinity for AtO+.However, density functional theory (DFT) calculations showed that S or N atoms are more likely to interact with the O atom in AtO+rather than directly coordinating with At atom.It also had been demonstrated that both At+and AtO+can form 1:1 and 1:2 complexes with Cl-,Br-, SCN-and thiocalix[4]arene tetrasulfonate [45,46].But there is still a lack of adequate evidence for the coordination between At and these selected ligands and the formations of corresponding complexes were more likely driven by ionic interactions.In fact,how to obtain enough experimental data to confirm the interaction between astatine and donor atoms may be the key and diffi-cult point of relevant research.
In recent years, computational chemistry which is not dependent on the availability of astatine, has become one of the important means to investigate At chemistry.For a lot of species including At-, AtX (X=H/H+, F-Ts), AtF3, AtO+, HOAt, CH3At and Y3C-At(Y=F to At),etc., the electronic property [47–54], bonding mechanism [55–59], solvation mode [60–64], halogen bond [65–71],molecular configuration [72–76] and adsorption energy [77] have been well studied.For organic astatine compounds, related researches mainly focus on the inherent mechanism of C-At bond[78–80] as well as the aromaticity of astatine-substituted borazine(B3N3At6) and benzene (C6At6) [81,82].It should be noted that for a heavy element like At, spin-orbit coupling and relativistic effects have very important effects on the theoretical calculations, so one must pay enough attention when talking about related bonding process.Meanwhile, for a certain characteristic property, different calculation methods may have huge difference in the final results.For example, for the first ionization energy and electron affinity of At, the calculated values by different methods vary in the range of 8.965~10.004 and 2.110~2.423 eV, respectively [47–49].Therefore, before experimental evidence is obtained, the accuracy of theoretical calculation will lack reliable calibration.Fortunately, by using laser ionization spectroscopy, the first ionization energy of At was determined by Rotheet al.[83], which is 9.31751(8) eV,while the electron affinity was identified as 2.41578(7) eV by Leimbachet al.[84].The determination of both basic data of At is of great significance.Firstly, it can offer important references which will prompt the computational researches involving At chemistry.Secondly, these data are helpful to recognize the reaction mechanism, especially for211At labeling and deastatination.Thirdly, the development of At study can facilitate the fundamental research on some other heavy elements like Ts.
2.3.Production of 211At
The production of radionuclide211At can be achieved by two approaches,211Rn-211At generator and209Bi(α, 2n)211At reaction on accelerator.211Rn has a longer half-life (t1/2=14.6 h), allowing211At to be supplied over medium and long distances.Even so, an great obstacle is that it uses 42/60 MeV of6/7Li to bombard natural Bi target for obtaining211Rn through209Bi(6Li, 4n)211Rn or209Bi(7Li,5n)211Rn reaction, which has strict requirements on accelerators with high energy of Li ions.Additionally,206Po,210At and210Po exist in the decay chain of210Rn [from209Bi(7Li, 6n)210Rn reaction]may contaminate211At products, which is a big challenge to separation technology.Therefore, the production of211At by209Bi(α,2n)211At reaction on accelerator is still the first choice at present.It is easier to achieve and has a high yield (16.3 to 41 MBq μA-1h-1).One thing should be paid attention to is that theαbeam energy must be strictly controlled below 29.0 MeV (usually 28.0~29.5 MeV) to avoid/minimize the amount of210At from209Bi(α, 3n)210At reaction (nonnegligible,>30 MeV).For current status in the production of211At, as well as how to break the logistical borders between the academic and commercial networks in the development of211At-labelled pharmaceuticals, one can turn to the latest review given by Fenget al.[85].
Generally, the most common separation of211At is achieved by high-temperature distillation.The basic principle is that the boiling point of At is 335 °C, while that of Bi is as high as 1564 °C.Therefore, the separation of211At from irradiated targets can be achieved by controlling the heating temperature.In a typical separation (Fig.2), the irradiated Bi target is placed in a furnace connected to a stable carrier stream, and is heated rapidly to 650~800°C to make211At sublimated.Gaseous211At will be carried out by N2, flow into the cold trap linked with negative pressure, condense and accumulate in the capillary.In the end, different solvents such as chloroform, methanol or NaOH are used to wash the capillary to obtain211At products.The whole separation process can be controlled within 30 min.The211At obtained by distillation is nearly carrier-free and there is no need to dissolve the irradiated target before separation.Hence, distillation is still the preferred approach used in the separation of211At.However, the recovery of211At in this approach is not stable and may vary in the range of 51%~83%[85].
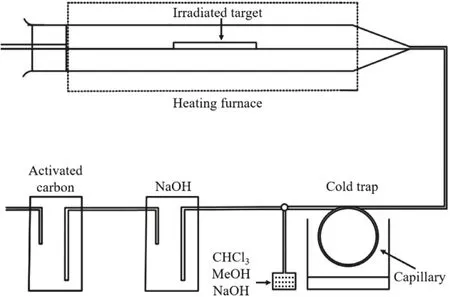
Fig.2.A simplified distillation apparatus for separating 211At.
In recent years, researchers also began to pay more attention to wet separation of211At again.Burnset al.[86] demonstrated that 3-octanone and methyl isobutyl ketone can effectively extract211At from nitric acid solution with Bi highly retained in water.Furthermore, by loading 3-octanone onto Amberchrom®CG300M resin, they found that the resulting impregnation resin can achieve high-performance separation of 151.7 and 362.6 MBq of211At [87].Specifically, the irradiated Bi target was first dissolved by 4~6 mol/L HNO3, and the obtained solution was then loaded onto the impregnation resin column.The Bi substrate and other impurity nuclides were removed by using 5.9 mol/L HNO3,while211At was retained on the column with a loss of<1%.Using ethanol as eluent, more than 90% of211At was washed from the resin, with the total volume lower than 1 mL.Woenet al.[23] proposed to use commercial pre-filter resin (100~150 μm) to recover211At from the irradiated Bi target.Different concentrations of HCl and NH2OH·HCl were used to remove Bi matrix and other impurity nuclides.211At retained on the column was eluted by 2 mol/L NaOH, with a recovery of 51%.Watanabeet al.[88] also tried to separate211At using anionic resin and found that211At would stay on the anionic resin when 8 mol/L HCl and HNO3rinsed the column, of which 75.3%~79.1% would be eluted by different concentrations of NaOH or NaOH-EtOH.For all the above-mentioned wet extractions, the separation time was in the range of 20~90 min,which is quite comparative to that of distillation.
There are also some reports of automatic extraction devices based on liquid-liquid or solid-liquid extraction [89,90].However,the specific activity of obtained products was not high enough,which was not conducive to211At labeling.Overall, the wet separation avoids using high-temperature devices, so there is no need to worry about the volatility of211At, and the operation is easier to achieve automation for large-scale production.However, target dissolution is more time-consuming, and the possible existence of residual Bi and impurity nuclides may influence the quality of211At product.For impregnation resin, the extractant may detach from the stationary and become impurity in the final product,whether this would influence211At radiolabeling still needs to be checked.Distillation and wet separation should complement with each other, and both are still under continuous development, especially the emergence of automatic separation device.How to improve the separation efficiency of211At will be one of the keys to promote the basic research and related radiopharmaceuticals.
3.211At-based radiopharmaceuticals
3.1.Labeling strategy of 211At
Usually, the radioastatination of various carriers can be achieved through either nucleophilic or electrophilic reactions.Unlike radioiodine,211At is difficult to bind directly to proteins or peptides to form stable radiolabeled compounds.Therefore, a bifunctional coupling agent which can bind with both the targeting carrier and211At is always needed.DFT calculations show that the stability of C–At bond is closely related to the chemical environment of At, which is also consistent with the change of C-I bond[78,79].Briefly, C–At bond gradually becomes more stable in the order of in alky<alkenyl<aryl compounds,i.e.,sp2C–At bond is stronger than sp3one and higher conjugation is helpful for stabling C–At bond.Interestingly, B–At and C–At bonds exhibit completely opposite polarities: At in C–At bond is positive-charged, while it is negative-charged in B–At bond.This can well explain why C–At is more vulnerable to nucleophiles.Relatedin vivoexperiments also showed that211At-labelled compounds linked by borane derivatives are more stable than those linked by aromatic ones [91–95],but the former may influence the targeting and circulation of the carriers, making corresponding radiopharmaceuticals show longtime retention in blood, kidney or liver [96–99].Therefore, the potential biotoxicity requires further systematic evaluation before its final application in211At labeling.
Common bifunctional coupling agents involving both C–At and B–At bonds for211At labeling are shown in Fig.3a, and more details about related radioastatination reactions have been described in other work [100].Some nontypical approaches, including coordination of calixarene [101]/metal-organic complex [102–105], clickchemical self-assembly [106], nucleophilic substitution using boric acid/borate ester [107–109] and nano-coprecipitation [110,111], are also alternative paths for211At labeling.However, due to the lack of reliable resultsin vivo, whether they can work in211At-labelled pharmaceuticals also needs further verification.Currently, one-step211At labeling based on aryl-trialkylstannane ester (ATE) derivatives is still the most widely-used strategy.In a typical process, ATE derivatives are first conjugated to the targeting vectorsviaamide reaction, resulting in corresponding precursors for halodestannylation (Fig.3b) [112].Recent studies have shown that the monoclonal antibodies (mAbs) with alkyltin-based prosthetic groups obtained in this method are highly stable at -20~4 °C for over 3 months in PBS (pH 7.4), which are still available for effective radioastatination[113].
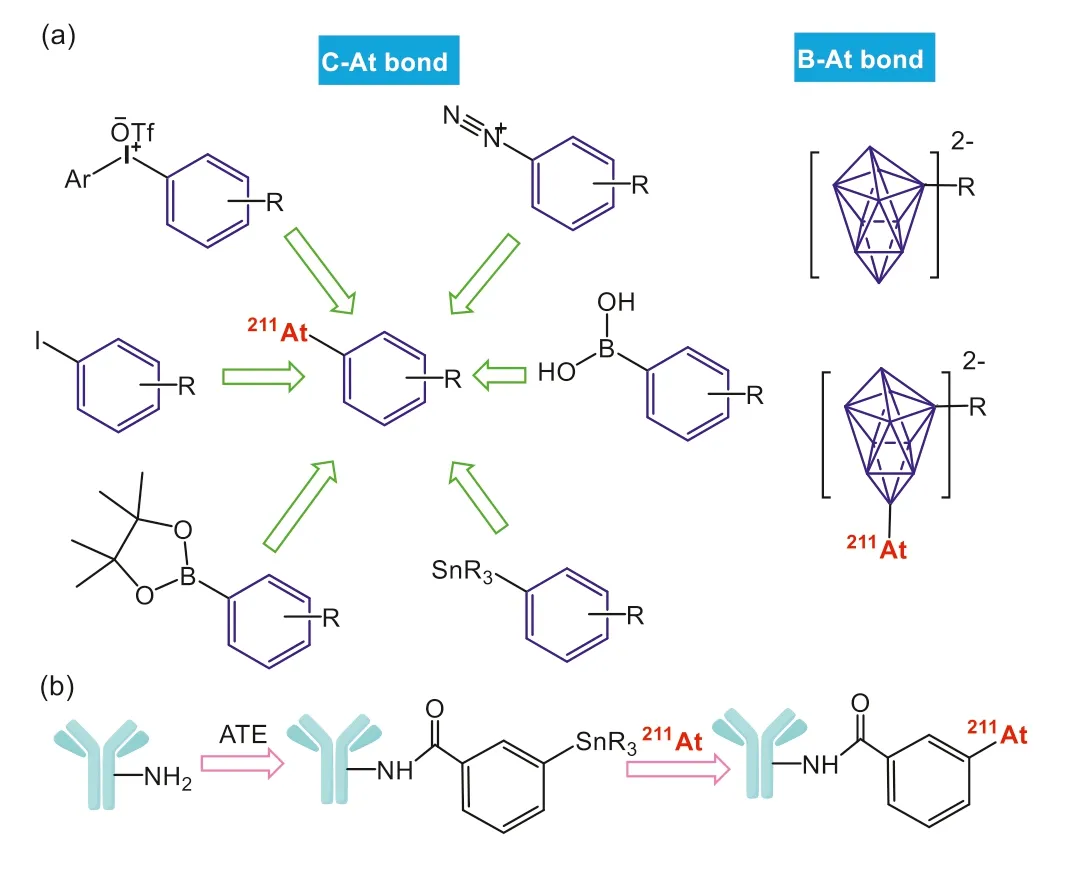
Fig.3.(a) Different bifunctional coupling agent used for 211At labeling and (b) the most common radiolabeling strategies in current radioastatinated compounds.The blue cartoons present targeting vectors.
In general, the instability of211At-labelled compounds is mainly attributed to the break of C–At bond.Theoretically, diminishing the electrical deficiency of At is an alternative way to consolidate the C–At bond.For example, incorporatingN-methyl or methylene between the amide bond and aromatic ring can significantly elevate the stability of corresponding labelled compounds [114,115].The basic principle is that the amide bond directly located on the benzene might increase the electrical deficiency of At, which is unfavorable for the stability of C–At bond.Of course, the bifunctional coupling agent and pharmacophore can also be linked by other approaches rather than amide bonds,e.g., reducing the disulfide bonds in proteins to sulfhydryl (-SH), which are then bonded to cycloolefin-containing bifunctional coupling agent through addition reaction [116].Although related radiolabeling compounds are less prone to occur deastatination [117], size exclusion highperformance liquid chromatography (SE-HPLC) analysis indicated that reductants can cause partial cleavage of related proteins, diminishing its purity and immune-activity [118].It is worth mentioning that211At can also be directly labelled on RGD peptides through the tributyltin pre-incorporated into the vectors [119].Corresponding labelled compounds keep high tumor targeting, suggesting their potential in211At-based TAT.
The introduction of corresponding substituents beside At is another way for improving the stability of labeling compounds.However, Talanovet al.[120] found that directly incorporating electrondonating groups such as methoxyl or methanethiol at theorthoposition of alkyltin tends to reduce the radiochemical yield without enhancing the stability of corresponding labelled compounds.One possible reason is the introduction of substituting groups may increase the steric hindrance for halodestannylation.On the contrary, guanidyl atortho- ormeta-position can significantly improve the radiochemical yield and the stability of211At-labelled compounds [121–124].Obviously, guanidyl with longer chain structure is favorable to reduce the electrical deficiency of At while avoiding extra steric hindrances in astatination.In addition, when conjugated with pyridine instead of benzene derivatives, the stability of corresponding211At labelled compounds also become lower [94],which may be due to the electron-drawing effect of N atom.
It should be noted that the211At labeling rate may decrease when the radioactivity increases from pre-experimentally to clinically required amount.One of the main reasons is that high doses of radionuclides can produce significant radiation effects on the whole system through two approaches,i.e., the solvent-induced consumption of bifunctional coupling agent and the redistribution of211At species.According to Pozziet al.[125], when chloroform was used as the reaction medium, ATE derivatives would undergo 50% degradation even at doses below 500 Gy.It was attributed to that chloroform tends to produce electrophilic chlorine radical under irradiation, which can consume ATE derivatives and reduce the labeling rate of211At onto the organic precursors [126].In methanol and benzene, more than 85% of the labeling precursors still remain stable even at the dose of 3500 Gy.The analysis results of HPLC showed that over 90% of211At exist as a single species in methanol when the dose was less than 1000 Gy.In this situation,the halodestannylation can easily be achieved.However, when the dose reaches 3000 Gy, At mainly exists in the form of At-, which cannot be effectively involved in electrophilic reaction to achieve radiolabeling [127].In order to address the conjugate of high activity211At onto targeting carriers, the most practical method is adding oxidants (such asN-succinimide orN-iodosuccinimide) in the reaction system, to inhibit the reduction of211At species [127–129].Besides, it is often necessary to perform large-scale211At radiolabeling before clinical trials, to verify the stability and reliability of the designed labeling strategies [130–133].
Actually, due to the limited availability of211At, labeling chemistry in a new system is generally carried out based on radioiodine (125/131I) before being applied to radioastatination [134–137].Typically, radioiodinated compounds are more stablein vivo, which is benefit from the better stability of C–I bond [138].As a result,compounds labelled with radioiodine and radioastatine may differ in biodistribution.For instance, Wilburet al.[139] found that the retention time of211At labelled compounds in kidney was significantly longer than that of125I-based conjugates, even the same targeting vectors were employed.Hence, it is always necessary to re-examine and compare some vital items, such as biodistribution and biotoxicity between radioiodinated and radioastatinated radiopharmaceuticals.
3.2.211At-based compounds in preclinical study
3.2.1.Na211At
Na+/I-symporter (NIS) is an integral plasma membrane glycoprotein most commonly associated with the thyroid gland that mediates active transport of I-to thyroid follicular cells [140].Because iodine is an important component of triiodothyronine and tetraiodothyronine, thyroid function and its systemic consequences depend on adequate uptake of I-.In addition to thyroid cells, NIS is also expressed in other tissues, including salivary glands, gastric mucosa, and lactating mammary glands.The ability of NIS to accumulate I-has therefore provided an important basis for targeted imaging and treatment of thyroid-associated diseases with radioiodine (mainly Na131I), which has achieved great clinical success.
The successful medical application of Na131I naturally encouraged researchers to employ Na211At in NIS-expressing tumors[141].First of all, there is a very high similarity between these two halogens, so, theoretically, NIS can also mediate211At-transport to thyroid or other related tumors [142–145].Secondly, dosimetry studies had shown that211At had the highest biological effect in thyroid gland when compared with60Co,99mTc,123I,125I and131I [146–151], resulting in significantly increased gene upregulation [152–155].Thirdly, the overall irradiation of the eyes caused by211At was nearly 5 times lower than that from131I irradiation [156].Unfortunately, although the growth inhibit effect of Na211At in NIS-expressing tumor models could be satisfactory[157,158], some studies also indicated that the non-specific uptake of Na211At in several important organs including stomach, salivary gland, lungs and spleen was significantly higher than that of Na125/131I, making the mean absorbed doses nonnegligible and may cause corresponding toxicity to organism [159–162].
Recently, Watabeet al.[10] found that one of the main reasons for this kind of biodistribution of free211At might be that astatine in aqueous solutions tends to exist in a plenitude of species including At+, AtO-, At(OH)2-, AtO2-, AtO(OH)2-, AtO+in addition to At-, thus reducing NIS mediated targeting.Therefore, if211At could be controlled in a single state, better therapeutic effect might be expected.Fortunately, it was found that the uptake of Na211At dispersed in citric acid solution by thyroid cancer cells were significantly increased.Correspondingly, more obvious tumor inhibition was observed in a dose-dependent manner.However, toxicity tests in both normal and tumor-bearing mice showed that 1.0 MBq of Na211At would cause weight loss, bone marrow growth inhibition,and pathological changes in subjects [163,164].Unfortunately, the application of Na211At in NIS-expressing tumors was overshadowed again.
3.2.2.211At labelled small molecules
Small molecules tend to show rapid tumor accumulation and normal tissue clearance, which is well matched with the halflife of211At.Considering thatαparticles can cause relatively limited cell damage due to short path length, small molecule with higher penetration may have some advantages in tumor therapy.Metaiodobenzylguanidine (MIBG) is a functional analogue of neurotransmitter norepinephrine and is able to bind with sympathetic medulla tissue by norepinephrine transporter (NET).NET is upregulated in many neuroendocrine tumors, which enables131I-labelled MIBG ([131I]MIBG) derivatives to be versatile in the treatment of neuroblastoma, pheochromocytoma and paraganglioma [165].It is not hard to image,211At-labelled benzylguanidine ([211At]MABG)derivatives are expected to show better performance in the treatment of NET-associated tumors [166–173].Boydet al.[174] illustrated that the inhibition of growth cancer cells transfected with methyladrenalin transporter (NAT) gene by [211At]MABG was much higher than that of [131I]MIBG.However, [131I]MIBG could result in bystander effect in solid tumor, which is less obvious forαemitter predominated endoradiotherapy [175].Recently, Ohshimaet al.[176] found that [211At]MABG could show significant antitumor effect in pheochromocytoma murine models.The tumor volume in the control group increased by 509.2% ± 169.1%, while those in the [211At]MABG administrated groups (0.56 MBq) was 9.6% ± 5.5% at 21 d post-injection.Although higher injection doses of [211At]MABG (1.1–3.3 MBq) would cause transient weight loss and leukopenia to subjects, but all mice regained weight at 28 d post-injection.However, the toxicity was irreversible when the dose was further higher than 4.4 MBq [177].
In order to maintain abnormal growth, several types of amino acid transporters are over expressed in tumor cells.This has facilitated the uptake and transportation of various amino acids in tumor cells, even crossing the blood-brain barrier.Therefore, various211At-labelled amino acid derivatives have become one hot topic in TAT.Meyeret al.[178] found that both[211At]-L-phenylalanine could specific bind to glioma cells.Antitumor studies in glioblastoma showed that both radioastatinated phenylalanine compounds could effectively inhibit glioma tumor growth and prolong the survival rate of murine models [179,180].Ohshimaet al.[181] demonstrated that [211At]-L-phenylalanine also showed the ability to suppress the tumor growth and prolong the survival of murine ovarian cancer models.Xieet al.[182] found that 4–211At-astato-N-[4-(6-(isopropylamino)pyridine-4-yl)-1,3-thiazol-2-yl]-N-methylbenzamide (211At-AITM) targeting ectopic metabolic glutamate receptors had good therapeutic effect in melanoma-bearing mice without obvious biotoxicity.In addition, Kaneda-Nakashimaet al.[183] also demonstrated that211Atlabelledα-methyl-L-tyrosine (211At-AAMT) could double prolong the survival of pancreatic cancer models and effectively reduce the lung nodules in B16F10 metastasis models.
Makvandiet al.[184] conjugated211At to 1-(4-iodophenyl)-8,9-dihydro-2,7,9a-triazabenzo[c,d]azulen-6(7H)-one, obtaining a radioastatinated compound [211At]MM4 which could inhibit tumor growth in murine neuroblastoma model.The median survival was increased from 66 days in the control group to 87 days in the treatment group received a twice-fractionated injection of 370 kBq of [211At]MM4.However, when a single injection of higher dose(1480 kBq) was administrated, significant weight loss was observed in murine models.When [211At]MM4 was used in combination with an immune checkpoint inhibitor, anti-programmed cell death protein 1 (anti-PD-1) [185], tumor growth was found to be 100%suppressed.The mechanism for the synergistic effect of radiotherapy and immunotherapy should be that the former can induce inflammation by causing DSBs inside cancer cells, and stimulate the organism to re-produce immune response.This is favorable to destroy the inhibitory effect of tumor cells on immune system,achieving efficient tumor treatment.
Kiesset al.[186] labelled211At to a small molecule inhibitor which can target prostate specific membrane antigen(PSMA), and obtained the radioastatinated (2S)-2-(3-(1-carboxy-5-(4–211At-astatobenzamido)pentyl)ureido)pentanedioic acid.The211At-labelled PSMA inhibitor was able to effectively inhibit tumor growth and prolong the survival of murine micro-metastatic models, but the uptake of radioactivity in thyroid and stomach was high, while the damage to the proximal renal tubules were also observed during the treatment.Different strategies had been adopted to address these two issues, including prolonging the chain linking pharmacophore and coupling agent, introducing guanidyl group atortho-position of211At, or changing the benzyl ring to pyridine [187].As a result, another four211At-labelled PSMA inhibitors were prepared, among which [211At]GV-620 had exhibit the highestin vivostability and shortest renal retention time.Measeet al.[188] also proposed that conjugating211At to high-performance177Lu-labelled pharmaceuticals might be another alternative approach to prepare new effective radiopharmaceuticals with potential in TAT.A series of211At-labelled compounds (177Lu was replaced by175Lu) targeting PSMA were prepared, of which211AT-3-Lu had shown excellent targeting ability, pharmacokinetic characteristics, good antitumor effect and almost negligible biotoxicity.
Recently, our group found that fibroblast activation protein inhibitors (FAPIs) featured with rapid metabolism and short retention in tumor might be more favorable targeting ligands for211At with relatively short half-life [135].211At-labelled FAPI-04 was synthesized for the first time with an ideal radiochemical yield and radiochemical purity [189].It was demonstrated that tumor growth was dramatically slowed by211At-FAPI-04 in a dose-dependent manner: glioma-bearing mice received 0.18, 0.37 and 0.55 MBq of211At-FAPI-04 showed a reduced tumor volume of approximately 55%, 70% and 76% on the 15thday, respectively.The median survival in211At-FAPI-04 group (24.0, 30.0 and 33.0 days for 0.18, 0.37 and 0.55 MBq of211At-FAPI-04, respectively) was significantly improved compared to that in saline group (16.5 days), causing no significant changes in body weight or toxicity in the kidney, liver,stomach and thyroid over the experimental period.
3.2.3.211At-labelled peptides
Peptides are compounds ofα-amino acids linked by peptide bonds, which are intermediate products of protein hydrolysis.The advantages of radiolabeled peptides include relatively simple chemical synthesis, available radiolabeling, rapid clearance from circulation, faster penetration, more uniform tissue distribution, and lower immunogenicity [190].Therefore, in recent years,211At-labelled peptides have become active in tumor therapy.Vaidyanathanet al.[191–193] was the first to realize211At labeling on octreotide derivatives and found that somatostatin receptor expressing tumor cells had certain uptake and endocytosis of the radioastatinated octreotide (211At-octreotide) derivatives.Zhaoet al.[194] found that211At-octreotide exhibited observable higher uptake in lung, spleen, stomach and intestines than in other tissues.Through histological examination,211At-octreotide demonstrated much more lethal effect than control groups (PBS, octreotide and free211At).
Aokiet al.[195] proposed to conjugate211At onto bombesin derivatives and performed the therapeutic effect of corresponding radiopharmaceuticals in prostate tumor models.It indicated that the prepared [211At]AB-3 had the highest labeling rate(28.2% ± 2.4%) when polyethylene glycol (PEG) chain was introduced between the bombesin scaffold and the labeling motif.[211At]AB-3 retained good affinity towards cancer cells and could be highly endocytosed, demonstrating that the whole radiochemistry process had no damage on the targeting of bombesin.However, endocytosed [211At]AB-3 tended to occur efflux over time and the uptake by tumor was as low as 2.56 ± 0.24 %ID/g, possibly originating from its unsatisfactory stability.
Our group [196,197] labelled211At onto a small molecule fusion peptide, VP2, and obtained a radiopharmaceutical [211At]At-SPC-VP2 targeting vasoactive intestinal peptide.It suggested that[211At]At-SPC-VP2 could effectively bind to a variety of tumor cells,including SW620 (colon cancer), A549 (human lung cancer), 4T1(mouse breast cancer) and 7721 (human liver cancer).In vivotreatment experiments showed that [211At]At-SPC-VP2 had high stability and could produce better anti-tumor effect than both nontarget nuclide and small molecule peptide.However, significant radioactivity was also observed in the stomach, which may be related to the deastatination of the labelled compoundin vivo.
3.2.4.211At-labelled antibodies
Recently, radioimmunotherapy (RIT) based on antibody has received increasing attention in preclinical and clinical researches[198].Naturally, conjugating211At to intact antibodies or related fragments to obtain astatinated pharmaceuticals with excellent anti-tumor effect is also one of the hotspots in TAT.Our group [199] found that211At-labelled mAb 3H11 and Fab fragments had obvious cytotoxic effect on human gastric cancer cells and211At-labelled Fab fragments showed higher tumor uptake(9.48~8.42 %ID/g) than211At-labelled mAb (~4.0 %ID/g).However,both radiopharmaceuticals had shown inadequatein vivostability.When211At was labelled onto insulin through pyridine derivatives,the obtained [211At]At-SPC-insulin showed goodin vitrostability and the purity of the radiolabeled compound was still higher than 95% for 24 h [200].The radioactivity was mainly accumulated in liver, with the uptake of 4.29 %ID/g at 30 min post-injection.However, thyroid, stomach, lungs and spleen also showed noticeable radioactivity uptake.
Kennelet al.[201] proposed to employ211At-labelled mAb 201B to treat BALB/C mice bearing approximately 100 EMT-6 lung tumor clones (approximately 2000 cells).They found that 185 kBq of211At-labelled 201B could extend survival rate of subject mice,and an injection of 370 kBq (25~40 Gy) of radiolabeled mAb was enough to eradicate all lung tumors, but mice received 740 kBq of211At-labelled 201B occurred pulmonary fibrosis at 3~4 months post-injection.Akabaniet al.[202] evaluated the efficacy of combination of211At- and213Bi-labelled 201B in lung tumor models by two-dimensional histological images and activity distribution at microscopic level.The results showed that radiolabeled mAbs were effective for the treatment of animals with small tumors, and the predicted therapeutic effect was consistent with experimental results.However, for large-size tumors, the therapeutic effect of radiopharmaceuticals was very limited.
Perssonet al.[203] found that211At-trastuzumab possessed a highly specific binding ability to breast cancer cells (KD=1.8 ±0.3 nmol/L) and 74 cell-associated decay of211At /cells could produce a cell survival fraction as low as (4.5 ± 0.8)×10-4.Akabaniet al.[204] pointed out that the relative biological effect of211At-trastuzumab was twice higher than that of external irradiation therapy, and it was able to inhibit the proliferation of HER2-positive tumor cells.Furthermore, the therapeutic effect of211Attrastuzumab on carcinomatous meningitis was verified in murine models [205].It had been proved that the median survival of tumor-bearing mice increased from 21 days in saline group to 45 and 48 days after intracranial administration of 1.22 and 2.44 MBq of211At-trastuzumab, respectively.When animals were inoculated with a lower tumor burden and received 1.70 and 3.40 MBq of211At-trastuzumab, the median survival was further extended to 68 and 92 days.An evaluation of radiotherapy efficacy of211At-trastuzumab in ovarian cancer models given by Palmet al.[206] further indicated that 500 μg cold trastuzumab combined with 400 kBq of211At-trastuzumab could achieve tumor ablation with no obvious biotoxicity occurring during the treatment.
Kodairaet al.[207,208] found that the anti-tumor effects of211At-trastuzumab on peritoneal metastasis of gastric cancer were significantly dependent on the injection method.The uptake of211At-trastuzumab by tumor was more than 60 %ID/g by intraperitoneal injection, while it decreased to 18 %ID/gviavein injection.Two of the six mice received 1.0 MBq of the radiopharmaceuticals achieved tumor ablation, while the growth of the tumor was significantly inhibited in other three models.Kodairaet al.[209] confirmed that intravenously-injected211At-trastuzumab was able to significantly accumulate in liver tumor tissues, and the absorbed dose was in the range of 1~7 Gy (peak at 2 Gy).Subsequently,211At-trastuzumab was demonstrated to have significant therapeutic effect on liver metastasis from a primary gastric cancer, and it could effectively prolong the survival of tumor-bearing mice without causing serious toxicity to murine models [210].Cederkrantzet al.[211] found that intraperitonially-injected211At-trastuzumab could result in an absorbed dose of 0~50 Gy for peritoneum in subjects, and a 34-week follow-up study indicated that the limiting dose should be 30 ~50 Gy.
Anderssonet al.[212,213] pointed out that intraperitoneal injection of 485~555 kBq of211At-MOv18 mAb could extend the median survival of ovarian cancer xenografted mice from 138 to 213 days, and 33% of the models were apparently tumor-free and possibly cured at 7 months.These positive results should prompt the application of211At-MOv18 in the therapy of ovarian cancer.However, in the past decades, the most widely-studied211At labelled compounds for treating ovarian cancer was based on MX35 F(ab’)2, a murine mAb fragment targeting Leyantigen.Bäcket al.[214] proved that the uptake of211At-labelled MX35 F(ab’)2by human ovarian tumor was 14 %ID/g at 7 h post-injection, while the ratio of tumor to blood uptake was 6.2 at 40 h post-injection.Elgqvistet al.[215,216] systematically evaluated the RIT efficacy of211At-labelled MX35 F(ab’)2in ovarian cancer models and found that tumor-free fraction (TFF) for mice bearing 4-week tumor were 25%, 22%, 50% and 61% after injected with 25, 50, 100 and 200 kBq of the radiopharmaceuticals, respectively.The minimum required activity of211At-labelled MX35 F(ab’)2for the treatment of ovarian cancer models was suggested to be 100 kBq, since TFF was higher than 50% when further increasing the dosage.A series of researches suggested that a variety of factors, including initial tumor volume [217–219], injection method [220,221] and specific activity of radiopharmaceuticals [222,223] had important impacts on the therapeutic effect of211At-labelled MX35 F(ab’)2ovarian cancer models.Briefly, high specific activity of211At-labelled MX35 F(ab’)2tends to exhibit better therapeutic effect on small-size tumors,and biotoxicity could be obviously diminished when fractionatedinjection is adopted.
The therapeutic effect of213Bi and211At-labelled MX35 in 2-week ovarian cancer models showed that TFFs were 0.60 and 0.90 for ~2.7 MBq of213Bi-labelled mAb and ~0.44 MBq of211At-labelled mAb, respectively [224], indicating that211At had the advantage of achieving better therapeutic effect.Chouinet al.[225] quantified the activity of211At-labelled MX35 F(ab’)2in micro-metastatic ovarian cancer models and found that high uptake and good retention of radiopharmaceuticals mainly existed on the surface of tumor cells, and 1.0 MBq of labelled compounds could make the tumor cells receive a dose of at least 12 Gy.For larger micro-metastatic tumors, activity uptake per cell was lower,probably due to the limited penetration of mAb.However, the average absorbed dose of tumor cells was still higher than 30 Gy due to the presence of crossfire.Bäcket al.[226] thus conducted a study of long-term renal toxicity of211At-labelled MX35 F(ab’)2in ovarian cancer model, and suggested that an average absorbed renal dose of ~10 Gy was acceptable and kidney would not be the primary dose-limiting organ.
In fact, as a murine mAb, MX35 may induce immune response in humans, limiting the possibility of subsequent fractional therapy trials.Therefore, Lindegrenet al.[227] investigated the differences in cell affinity and targeting specificity between211Atlabelled humanized mAb (211At-Rembab200) and211At-labelled MX35.They found that both radiopharmaceuticals possessed high similarity in terms of tumor antigen and cell binding, confirming that211At-Rembab200 also had potential in the treatment of ovarian cancer.Palmet al.[228] also radioastatinated humanized farletuzumab (211At-farletuzumab) targetingα-folic acid receptor, for the treatment of abdominal metastatic ovarian cancer.In murine disseminated ovarian cancer models, they demonstrated that211Atfarletuzumab was able to inhibit the tumor growth more effectively than211At-labelled MX35.This might be due to that farletuzumab has a weaker affinity to ovarian cancer cells than MX35,allowing corresponding labelled mAb to penetrate the tumor more effectively.
211At-labelled mAbs have also received much attention in TAT for various blood diseases [229].Wesleyet al.[230] found 16.65 MBq/kg of212Bi- and 2.22 MBq/kg of211At-labelled humanized mAb anti-Tac (211At-HAT) could prolong the survival of cynomolgus cardiac allograft models to 14.0 ± 1.3 and 26.7 ± 2.4 days,respectively (8.2 ± 0.5 days in the control group), indicating that211At behaved better in tumor inhibition.A radioastatinated anti-CD25 mAb,211At-labelled 7G7/B6, had also shown potential for leukemia treatment and 555 kBq of211At-labelled 7G7/B6 could significantly prolong the mean survival time of karpas299 leukemia models from 24 to>76 days, with 5 of the 15 mice living more than 5 months [231,232].Moreover, a combination therapy in Tcell white blood models using211At-labelled 7G7/B6 and cold daclizumab made 91% of subjects live as long as 94 days, which was 36% and 50% in211At-labelled 7G7/B6 and cold daclizumab groups [233].Nakamaeet al.[234] found that211At-B10–30F11 targeting CD45 was high-performance in generating bone marrow suppression, since all mice administrated with 0.74 or 1.85 MBq of radiopharmaceuticals had lethal myeloablation.Orozcoet al.[235] demonstrated that211At-B10–30F11 could significantly prolong the median survival time of leukemia murine models in a dose-dependent manner: the median survival time was 123, 101,61 and 37 days for subjects received 888, 740, 444 and 0 kBq of211At-labelled 30F11, respectively.
In canine model, Chenet al.[236–238] also demonstrated that seven of the eight models would exhibit long-term donor monocyte chimerism (19% to 58%) when pre-treated with 5.735~23.125 MBq/kg of211At-CA12.10C12 before hematopoietic cell transplantation (HCT) followed by short-term immunosuppression of cyclosporine and mycophenolate.At doses below 14.985 MBq/kg,the dogs exhibited transient hepatotoxicity and subsequently autogenous recovery, without nephrotoxicity observed at the end of an 18~53 weeks follow-up.It has been demonstrated that a cell dose exceeding 1×106cells/kg in combination with nonmyeloablative doses (14.80~18.13 MBq/kg) of211At-CA12.10C12 could sustain the engraftment level in dog models after autologous HCT.A subsequent therapeutic study in canine models demonstrated 6.956~14.319 MBq/kg of211At-CA12.10C12 in combination with 9.2 Gy total body irradiation (TBI) appeared to be more effective to overcome graft rejection in red cell disorders and other nonmalignant conditions treated with allogenic transplantation [239].
Petrichet al.[240] found that211At-AntiCD33 had significantly higher specific binding ability to leukemia cells and could effectively inhibit their proliferation.In vivostudy on murine models showed that211At-AntiCD33 possessed a characteristic biodistribution consistent with non-specific antibody retention in the reticuloendothelial system [241].The largest proportion of radioactivity was observed in blood and blood-rich tissues, which was still over 85% at 21 h post-injection.During a fellow-up of 6 months,no significant toxicity occurred in all treatment group.O’Steenet al.[242] carried out the treatment of invasive multiple myeloma based on211At-OKT10-B10 and found that the radiopharmaceutical could effectively inhibit tumor growth and double the survival time of the model mice (>150 days).Gouardet al.[243] synthesized211At-labelled 9E7.4 and confirmed its therapeutic effect in multiple myeloma model: the survival rate at 150 d was 65% in the presence of 740 kBq of related radiopharmaceuticals.
Nestoret al.[244,245] conducted the evaluation of211Atlabelled U36 chimeric mAb on human neck squamous cell carcinoma (HNSCC) and found that the prepared radiopharmaceutical had high specific binding and endocytosis ability to cancer cells.The survival rate of each cell was estimate to be 10% after 50 decays, and 200 kBq of the radionuclides could prevent 90% of the tumor-bearing mice appearing tumor proliferation.Robinsonet al.[246] carried out the treatment of human breast tumors using211At-labelled SAPS C6.5, a bispecific antibody targeting HER2.It demonstrated that 0.740 and 1.665 MBq of211At-labelled SAPS C6.5 extended the survival time of tumor-bearing mice by 30 and 57 days, respectively, and the higher activity could occur 60% tumor ablation after 1 year.
Turtoiet al.[247,248] found that human lymphocytes could show high sensitivity of gene expression and rapid regulation after exposure to211At.Greenet al.[249] compared the anti-tumor effi-cacy of211At-labelled 1F5 mAb ([211At]1F5-B10) in large-size lymphoid xenograft and minimal residual disease (MRD) models.They found that [211At]1F5-B10 had a moderate effect on solid tumors and mice received 1.776 MBq of the radiopharmaceuticals lived only 2~3 times longer than those in the control group.In contrast, 70% of the MRD models received 555 kBq of [211At]1F5-B10 could achieve complete tumor elimination.Bäcket al.[250] confirmed that two-fractionated of 1.5 and 1.9 MBq of211At-labelled anti-PSCA A11 minibody (211At-A11) targeting prostate stem cell antigen could inhibit tumor growth by nearly 85%.Erikssonet al.[251] demonstrated that 2.5 or 5.0 MBq of211At-labelled BR96(chimeric mouse/human IgG1 mAb) could facilitate half of the murine colon cancer models to achieve tumor ablation, when the initial tumor diameters were ~10 mm.Although RIT induced transient weight loss and bone marrow toxicity, the overall survival of the mice was significantly better than that of the control group.
Liet al.[252] tried to conjugate211At to major histocompatibility complex CLASS I chain associated proteins A and B (MICA/B)and found the obtained211At-anti-MICA/B Ab had high binding ability to human osteosarcoma cells SaOS2 and U2OS, of which 1.0 MBq could effectively inhibit the growth of HCT116 p53-/--positive tumor.They also compared the anticancer efficacy of211At- and90Y-labelled anti-frizzled Homolog 10 (FZD10) in synovial sarcoma models [253].The results indicated that 1.85 MBq of the prepared211At-labbeld OTSA101 suppressed tumor growth immediately after injection, whereas this effect required several days in the case of90Y-labelled OTSA101.The combination ofα- andβ--emitters based radiopharmaceuticals was supposed to be more conducive in RIT.However, a study performance by Erikssonet al.[254] indicated that administrating 5 or 10 MBq/kg of211At-labelled BR9 at 25 d post-injection of 400 MBq/kg of177Lu-labelled BR96 had no effect on reducing the proportion of colon carcinoma bearing mice developing metastases.On the contrary, obvious bone marrow toxicity was observed in the treatment group.These results were discouraging, and at least proved that sequential RIT alone could not achieve synergistic effects.
For211At-based RIT, how to enhance the stability of211Atlabelled mAbs also drew much attention, and an available approach might be introducing adjuvants into the media.Sundberget al.[255] demonstrated that using lysosomal weak bases could improve the retention time of radiolabeled epidermal growth factor(211At-EGF) in tumor cells.Takashimaet al.[256] proposed to dissolve211At-anti-TF mAb in sodium ascorbate (SA) and found that 1.2% SA could significantly enhance the cellular binding of211Atanti-TF mAb from 2.67% ± 0.40% to 17.52% ± 3.58%.Compared with 1 MBq of211At-anti-TF mAb in PBS, those containing 1.2% SA could further prolong the median survival of gastric cancer models from 30 to 34 days.These results showed that reductive adjuvants might effectively improve the stability of astatinated compounds, resulting in better therapeutic effects.
Many preclinical studies have shown that mAb-based TAT can achieve tumor ablation when the maximum tumor radius was in the range of 0.1~0.2 mm, while it has little effect on tumors with larger size, which might be due to the limited penetration ability of mAbs.Hence, pre-targeting radioimmunotherapy (PRIT)has become attractive in TAT.A pre-targeting therapy can be divided into two steps: in the first step, the pre-targeting molecule(mainly mAbs) is injected into the organism.The second step is activation step, that is, after the targeting precursor has highly enriched at the tumor site, the effector molecule with good penetration ability is injected, which will be fixed by coupling with the pre-targeting molecule.Pre-targeting location can make the radionuclides better target the tumor, minimizing the biotoxicity of radionuclides to normal tissues.Wilburet al.[257] carried out a preliminary study of213Bi- and211At-labelled streptavidin for mAb pre-targeting.However, the effector molecules labelled with211At were not suitable for PRIT due to their poor stability or low labeling rate.Frostet al.[258,259] proposed to employ avidinconjugated MX35 (Avidin-MX35) as the pre-targeting molecule and211At-labelled biotinylated poly-L-lysine (211At-B-PLSUC) as the effector molecule to achieve PRIT.In micro-metastatic ovarian cancer models, they found that PRIT exhibited no significant advantage in final anti-tumor effect over common RIT [260].Navarroet al.[261] adopted five different click-chemical reaction strategies to conjugate the pre-targeting and effector molecules (polypeptides).Although BCN/TZ-IEDDA and DIBAC/N3-SPAAC showed obvious kinetic advantages, relevantin vivoexperimental results are still needed.
Severalin vitrostudies also have shown that [262–265], multiple211At-labelled mAbs (cocktails) or single211At-labelled mAbs in combination with unlabeled mAbs could inhibit tumor growth more effectively than single administration.However, due to the lack of relevantin vivoevaluations of anti-tumor effect, or evenin vivodistribution study, whether these strategies can work in the treatment of solid or metastatic tumors still needs to be verified.Actually, even for211At-labelled mAbs or related fragments of which the stability and biodistribution in tumor-bearing mice had been performed [266–273], the practical anti-tumor and long-term biotoxicity also need to be verified.
3.2.5.211At-labelled nanomaterials
The application of nanotechnology in human clinical practice is known as nanomedicine and offers a powerful tool with high potential and multiple applications.For211At-based endoradiotherapy, there are also some attempts based on nanomaterials.Kuckaet al.[274] achieved211At radiolabeling onto PEGylated silvercontaining nanoparticles, and found that, in the absence of reductant, the radiochemical yields could be over 94%.Moreover, the obtained211At-labelled nanoparticles exhibit high stability even when treated with a large surplus of competitive chloride ions.Hartmanet al.[275] also performed211At labeling on ultra-short single-wall carbon nanotubes (US-tube) in the presence of chloramine-T/Nchlorosuccinimide with radiochemical yields of 60.7% and 55.4% in water and methanol, respectively.The prepared211At-labelled UStube showed good stability, with the retention of211At on the nanotube kept as 72%~85% and 85%~93% after washed with PBS and serum twice, respectively.
Dziaweret al.[276] carried out the labeling of211At on gold nanoparticles (AuNPs) and performed the preliminary biotoxicity study of related radiolabeled nanomaterials.Specifically, neuropeptide fragment P(5–11) targeting the nerve type 1 receptor was first conjugated to 5~15 nm AuNPs, of which halogenation were achieved by subsequently adsorbing211At-.The prepared211At-AuNP-S-PEG-SP(5–11) had good stability and could inhibit the growth of glioma cells.Sporeret al.[277] found that mixing211At and 25~50 nm AuNPs before PEGylation could effectively achieve the211At labeling onto the gold-based inorganic vector with a radiochemical yield of 99%.In vivostudies indicated that intravenously-injected211At-labelled AuNP showed low uptake in the stomach and thyroid, high uptake in liver and spleen with long-time blood circulation.
Katoet al.[278] conjugated211At with AuNP-S-mPEG ranging from 5 nm to 120 nm AuNPs, and confirmed that the obtained radiolabeling nanomedicines could significantly inhibit the growth of glioma and prostate cancer cells.Furthermore,211At-AuNP-S-mPEG could well be located inside the xenograft after intratumoral injection.The tumor inhibition of211At-AuNP-S-mPEG was significantly related to the particle sizes, and 5 nm211At-AuNP-S-mPEG showed the best therapeutic effect, with no significant increase of the tumor size at 40 d post-injection.This might be that the smaller size of the nanomedicine was better favorable to penetrate to the center of the tumor.Liuet al.[279] also achieved211At labeling of gold nano-stars (GNS) with high radiochemical yield (>95%).The prepared211At-labelled GNS had shown excellentin vitroandin vivostability and could well inhibit tumor growth on glioblastoma multiforme (GBM) models.During a 14-day fellow-up, the average tumor volume in the control group increased rapidly from 82 mm3to 1456 mm3while that in the treatment group received 1.11 MBq of211At-labelled GNS increased slowly from 78 mm3to 189 mm3.These results suggested that211At-labelled GNS had great potential in the treatment of GBM.
Shiet al.[280] synthesized a semiconductor polymer nanopore(SPN) which was further modified with glucose-dependent insulinstimulating polypeptide (GIP).After radioastatination, a new nanomedicine211At-MeATE-SPN-GIP was obtained, of which 1.1 MBq could significantly inhibit tumor growth in pancreatic cancer murine models without weight loss, renal and thyroid function disorder occurring.Biodistribution showed that the radioactivity in the stomach was high, suggesting that the stability of211At-MeATE-SPN-GIP should be further enhanced.In addition, like all other nanomaterials, the uptake of intraperitoneally-injected211At-MeATE-SPN-GIP by liver, spleen, and lungs was still significant,which made the long-term toxicity to these organs need to be verified.
Compared with small molecules, peptides and mAbs,211Atlabelled nanoparticles are not well studied.There are many factors for this, including the inconsistence of half-lives between211At and the nanoparticles, the poor stability of nanoparticles, the uptake of nanomedicines by the reticuloendothelial system, and the longterm toxicity of nanoparticles,etc.Of course, some of the problems,such as the interception by reticuloendothelial system is an inherent issue for all nanomedicines, but not featured by211At-labelled nanoparticles.An alternative solution to this problem, at least for211At-labelled nanoparticles, is adopting local administration in tumor endoradiotherapy, with an attempt to reduce the metabolism of nanomedicinein vivo.
3.3.Clinical study of 211At-based radiopharmaceuticals
Up to now, there are several clinical trials involving211Atlabelled pharmaceuticals, which were driven by previous positive results of preclinical results.Akabaniet al.[281] found that211Atlabelled ch81C6 mAb could specifically accumulate in GBM tissue and the radioactivity was approximately 5 times higher than that in normal brain tissue.As a result, a lower survival probability of GBM cells than normal brain endothelial cells had been observed.Subsequently, a clinical study was carried out [282], in which 18 patients with recurrent brain tumors were injected with 71~347 MBq of211At-labelled ch81C6 through a surgically created resection cavity (SCRC) and subsequently subjected to remedial chemotherapy.It had been demonstrated that a total of 96.7% ± 3.6% of211At decayed in SCRC, and the mean blood pool percentage injection dose was ≤0.3.Six patients developed grade 2 neurotoxicity within 6 weeks of211At-labelled ch81C6 administration, which eventually disappeared completely in all but one patient.There was no grade 3 or higher toxicity observed in all patients, and no one required repeated surgery due to radionecrosis.Median survival was 54, 52, and 116 weeks for different patients with GBM, anaplastic astrocytoma and oligodendroglioma, respectively.
Anderssonet al.[283] conducted a phase I clinical study with211At-labelled MX35 F(ab’)2in 9 patients with ovarian cancer.All subjects were received 22.4~101 MBq/L of211At-labelled MX35 F(ab’)2viaperitoneal catheters.It indicated that the radiopharmaceuticals in the peritoneal fluid decreased to 50% initial activity concentration at 24 h, while those in serum increased to 6% at 45 h.Radioactivity in the thyroid would increase to 127% at 20 h without blocking, which was<20% with blocking.The cumulative urinary excretion was 40 kBq/(MBq/L) at 24 h, and the estimated absorbed doses were 15.6 ± 1.0, 0.14 ± 0.04, 0.77 ± 0.19, 24.7 ± 11.1 and 1.4 ± 1.6 mGy MBq-1L-1) for peritoneum, red bone marrow,bladder wall, unblocked thyroid and blocked thyroid, respectively.It has been demonstrated that bladder, thyroid, and kidney had the highest contribution to estimated absorbed doses, while lungs,stomach, and bladder shared the largest doses when tissue-weight factor was taken into account [284].The organ equivalent dose was 10% lower than the estimated tolerance dose when 100 MBq/L of211At-labelled MX35 F(ab’)2was administrated.
Hallqvistet al.[285] performed a phase I clinical study involving 12 patients with recurrent ovarian epithelial carcinoma who had developed secondary or complete resistance to chemotherapeutic agents.They found that when the dose of211At-labelled MX35 F(ab’)2was up to 215 MBq, no significant biotoxicity was observed during the treatment.Actually, most of the low-level biotoxicity was not directly related to endoradiotherapy itself.Four patients survived longer than 6 years, and one did not have tumor recurrence.Median overall survival was 35 months, with 1, 2, 5 and 10-year survival rates of 100%, 83%, 50% and 25%, respectively.Dosimetry studies showed that lower specific activity was associated with lower single cell dose, while in microtumors, higher specific activity might lead to lower central dose.No obvious radiotoxicity was observed during a 12-year follow-up, and none of the subjects showed any signs of decreased tolerance to recurrent therapy.
Based on the positive effects of211At-labelled mAbs in blood disease models, Liet al.[133] recently announced an impending clinical study (NCT03128034) for the treatment of advanced hematological malignancies.Meanwhile, O’Steenet al.[242] also planned to apply211At-OKT10-B10 in a clinical trial of treating invasive multiple myeloma.Absolutely, all these positive results will promote the clinical application of211At-based radiopharmaceuticals.
4.Challenges and prospects of 211At-labelled pharmaceuticals
As summarized in Table 3,211At-based radiopharmaceuticals have received increasing attention from 2000.However, the development of211At-labelled compounds is facing several challenges:

Table 3 Preclinical investigations and clinical trials of 211 At-radiopharmaceuticals in 2000-2022.
(1) Production of211At.Although some accelerators are currently capable of producing GBq-scale211At (up to 9 GBq)[85], there are currently no more than 30 in the world available for producing211At.Therefore, it is predictable that availability of211At will still be a problem in the future.Since the cost of a large accelerator is often high, the intervention of governments may be an important way to solve the problem of211At production and supply.Encouragingly,in several parts of the world, including the United States, the European Union and Japan, government agencies are taking concrete actions to build, upgrade or retrofit accelerators to increase the capacity of211At production [85].
(2) Basic chemistry of At.Compared to otherα-emitters, the recognition about the fundamental properties of211At is limited.At present, many fundamental researches involving At chemistry are still focused on the properties of electron configuration, species states and halogen bonds in simple systems, those based on211At labelled compounds are very rare.The exact mechanism of halogenated and deastanization of211At, the specific denaturation path of radioastatinated compoundsin vivo, and the law of C–At bond stability,etc., all lack thorough researches.Fortunately, the development of both computational chemistry and experimental methods have provided powerful tool to explore these is-sues, which will result in more great progress in the near future.
(3)211At labeling strategies.Although there are a lot of strategies for labeling211At, low reproducibility of radiochemical yield and poor stability of labelled compounds are still one of the important problems restricting the development of211At in TAT.How to find the effective approaches for conjugating211At and designed vectors, or how to find a more high-performance bifunctional coupling agent, will still be one of the key topics in future.Inspiringly, the nucleophilic substitution reactions based on aryl borate [107] and aryl iodobenzene [286] provide important references for the development of211At labeling and deserve to be tried in more studies.The developments of computational chemistry can also offer important guidance for the design and synthesis of new bifunctional coupling agents.
(4) Ideal targeting carrier.Because211At has a relatively short half-life, small molecules with shorter biological half-lives,including amino acids, inhibitors, peptides and so on, often seem to match211At better.However, after radioastatination, their specific targeting, stability and retention time at tumor site are not satisfactory.As for mAbs, they can show higher stability and retention time in tumor.However, their long biological half-lives are difficult to match the physical half-life of211At.In this situation, mAb fragments seem more favorable for211At-based radiopharmaceuticals.To avoid long-timein vivocirculation and the uptake by normal organs and tissues, local administration of211At-labelled mAbs/fragments might also be available to achieve regiontargeted radiotherapy.The selection of appropriate tumor type should be another option.As least,211At-labelled mAbs might be conducive to treat blood diseases due to their long-time circulation.Clearly, micro-metastatic tumors with a smaller size also seems to receive better therapeutic effect than solid ones.
5.Conclusion
In the past decades,211At-based radiopharmaceuticals have made great progress in basic physicochemical properties, labeling strategies, preclinical and clinical studies.Accurate determination of the first ionization energy and electron affinity can not only promote the recognition of At element itself, but also provide important reference standard for the development of computational chemistry.Clarification of At species in aqueous solution, especially in oxidation conditions, is of great significance for the experimental development of211At radiochemistry.New labeling strategies will give more and more choices for conjugating211At onto the targeting vector.211At-labelled radiopharmaceuticals have become more and more active in nuclear medicine, depending on the emergence of novel targeting vectors.Although there are still several challenges in the development of211At-related radiopharmaceuticals, the great successes in preclinical and clinical researches have already lighted up the prospect of211At in practical tumor radiotherapy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are grateful for the financial supports from the National Natural Science Foundation of China (No.22006105), the China Postdoctoral Science Foundation (No.2020M683309), and the Fundamental Research Funds for the Central Universities.
 Chinese Chemical Letters2022年7期
Chinese Chemical Letters2022年7期
- Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent development in selective Tau tracers for PET imaging in the brain
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging
- Radiolabeled peptide probe for tumor imaging