
Radiolabeled peptide probe for tumor imaging
2022-07-09 02:14YaQiongYanHaoWangYuliangZhao
Chinese Chemical Letters 2022年7期
Ya-Qiong Yan, Hao Wang, Yuliang Zhao
CAS Center for Excellence in Nanoscience, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China
ABSTRACT Radionuclide imaging is now the premier imaging method in clinical practice for its high sensitivity and tomographic capability.Current clinically available radio imaging methods mostly use positron-emission tomography (PET) and single-photon emission computed tomography (SPECT) to detect anatomic abnormalities that conventional imaging techniques typically have challenges for visualizing.Contrast agents are indispensable for radionuclide imaging, and the radionuclide is always attached to a suitable vector that achieves targeted delivery.Nowadays, peptides have attracted increasing interest in targeting vectors of contrast agents, mainly due to their high specificity for target receptors at nanomolar concentrations and low toxicity.Radiolabeled peptide probes as kinds of PET/SPECT tracers had become essential tools for clinical radionuclide diagnosis.This review mainly summarizes radiolabeled peptide probes for bioimaging, including fundamental concepts of radiolabeled peptide probe design, some typical peptide analogs radiocontrast agents for PET, SPECT, and the combination imaging.
Keywords:Peptide Positron-emission tomography (PET)Single-photon emission computed tomography (SPECT)Bioimaging
1.Introduction
Radionuclide imaging facilitates whole-body and non-invasivein vivotracking by sensitive clinical imaging techniques.It is a non-invasive imaging technique that could visualize biological processes in living subjects [1].The most available radionuclide imaging technologies are positron-emission tomography (PET) or singlephoton emission computed tomography (SPECT).Various diagnostic radionuclides were applied as contrast agents, including123I,99mTc, and111In for SPECT imaging and68Ga,18F and64Cu for PET imaging [2].The tomographic imaging of PET was reconstructed by detecting the two gamma rays with an energy of 511 keV in opposite directions [3].PET imaging takes advantage of the unique properties of radioactive isotopes that decayviapositron emission.SPECT imaging detects gamma-ray photons emitted during their radioactive decay by rotating the camera around the subject to capture the gamma emissions in 3D.PET offers higher spatial resolution, while SPECT has the advantages of more readily available,longer-lived and cheaper radioisotopes [4].PET and SPECT imaging have a clear diagnostic advantage with the most sensitive molecular imaging technique at the picomolar range [5].The development of SPECT and PET imaging, combined with specific targeting radiolabeled molecules, has opened a new era of clinical imaging [6].However, PET and SPECT have a crucial disadvantage, the lack of spatial resolution.Accordingly, SPECT/PET imaging is often combined with computed tomography (CT) or magnetic resonance imaging (MRI) for their high spatial resolution [7,8].
An essential goal of agent-based non-invasive imaging is the specific accumulation of an agent at the disease site [9].Targeted molecular imaging techniques have become essential tools to provide accurate and specific disease information.Unlike CT/MRI imaging, a suitable contrast/imaging agent is indispensable for PET and SPECT imaging.Since that, accurate targeting is crucial to obtain meaningful diagnostic information, especially for radionuclide imaging.Highly-quality radionuclide imaging requires radiotracers efficiently and highly enriched at specific sites.Accumulation of PET/SPECT tracers provides a way to image the molecular signature of disease [10].Sensitive and specific targeted vectors have attracted considerable research interest to meet the clinical needs of radionuclide imaging [11].Targeting vectors (e.g., peptides, nucleotides, antibodies, nanoparticles) dictate the biodistribution of the radioactive molecule.These targeting vectors are designed to accumulate at disease sites based on the difference in the lesion location in contrast to tissues.Promising targeting vendors display high-affinity binding to the specific receptors that are highly expressed on target cells and can be rapidly cleared elsewhere.
Peptides have many advantages, such as superior specificity and flexible chemical modifications [12].Peptide probes enable high affinities and specificities for their targets and fast clearance from non-target sites, resulting in excellent target-to-non-target ratios[13].They have modest stability and remarkable tissue/tumor penetration capability with lower immunogenic effects than antibodies [14,15].Since then, peptide probes have garnered increasing attention as a targeting ligand due to facile synthesis, lower toxicity,and rapid and specific accumulation at target receptors [16,17].The use of solid-phase peptide synthesis and phage display libraries have led to rapid high throughput synthesis and screening of peptide imaging agents [18].The specific sequences of targeting peptides play an essential role in designing peptide contrast agents for bioimaging [16,19].Moreover, unbound peptide contrast agents are rapidly clearedin vivoapplications so that radiolabeled peptide probes had low background uptake in non-target tissues [20,21].Therefore, peptides became a promising candidate for radionuclide labeling.
Radionuclides have been used in diagnosis since the late 1950s[22].There is a growing wealth of literature on the topic.Radionuclide was divided into two broad classes: organically derived(11C,13N,15O,18F,76Br,124I), and radiometals (e.g.,64Cu,66Ga,68Ga,96mTe,111In) [23].Radionuclide labeling on targeting vectors is primarily important in practical applications [24].Generally, the radiolabeled peptide probe for bioimaging can be divided into three parts: radionuclide, chelating agents/linker, and targeting vector[20].There are two major approaches for radionuclide labeling: direct labeling and coupling to chelating agents.Chelating agents are used to effectively deliver radionuclidein vivofor imaging without any radiometal loss.Bifunctional chelators (BFCs) are a ubiquitous part of radiolabeled peptide probes.Bifunctional chelators are more suitable in practical application since they contain chelating ligands with reactive functional groups that allow them to be covalently attached (conjugated) to biologically relevant vectors [25].
Many peptide contrast agents have been developed for preclinical or clinical application (Fig.1).The following review presents an overview of radiolabeled peptide probes for bioimaging, including radionuclide labeling methods, typical peptide analogs for radiocontrast agents, and combination imaging of radionuclide imaging with other imaging modalities used for preclinical and clinical applications, would be summarized.
Fig.1.Representative types of radiolabeled peptide contrast agents for radionuclide imaging.
2.Peptide contrast agents for radionuclide imaging
2.1.Design of peptide-based radiocontrast agents
To radiological imaging a certain lesion, one needs to identify and synthesize a radiolabeled imaging agent.In radionuclide diagnostics, peptides usually play a significant part of the target vector to a specific biological site [26].The two key design considerations are (i) which radionuclide and targeting peptide should be use and (ii) how to conjugate the radionuclide and the targeting peptide.The characteristic of the radionuclides and the targeting peptides must be considered.Radionuclides and the selected targeting peptide should be safe and stable.Different properties such as half-life, decay mode,and biological response must be considered in advance.For example, iodine radionuclides offer different isotopes to perform medium-term (123I,t1/2=13.3 h) or long-term imaging studies(125I,t1/2=60.5 d) and even radiotherapy (131I,t1/2=8 d) with the same molecule.In order to avoid the isotopes detached from the host peptide or contrast agents during the circulation, the radiolabeling progress should be efficiently and physiologically stable but not be too stringent.As for the conjugation methods,it usually added chelate agents as a linker.The radionuclide is attached to the targeting peptide as a complex with a suitable chelator, frequently through an additional linker.There are plural factors that affect the creation of chelation architectures, including the choice of chelating agents and the additional functional linkers,the impact of the radiolabeling on peptide-receptor interaction,and the overall conformational structure radiotracers.Incorporating a radioisotope, chelator, and additional functional linkers like a spacer to a targeting peptide may cause significant changes to their physical properties [27,28].The most applied chelating agents are 1,4,7,10-tetraazacyclodecane-1,4,7,10-tetraacetic acid (DOTA), 2,2′,2′′-(1,4,7-triazacyclononane-1,4,7-triyl)triacetic acid (NOTA), diethylene triamine pentaacetic acid (DTPA), triethylenetetramine (TETA), and 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A) (Fig.2A).The radionuclides may hamper the interactions between the targeting peptide and their related peptide receptor, leading to the nonspecific distribution of the radiocontrast agentsin vivo.It has been reported that the high-affinity binding of Ln(III) ions with certain peptide sequences could interfere with the complexation of the peptide-DOTA conjugate.Conjugation of DOTA to the peptides as well as minor modifications of the peptides can affect binding affinity.The additional functional molecules like flexible linker or spacer polyethylene glycol (PEG) can alter the overall charge and hydrophilicity of the radiocontrast agents [29,30].
Various techniques have been developed that allow efficient radiolabeling of peptides.The ideally radiotracers labeling process should be selectivity, high efficiency, rapidity, and modularity.The peptides coupled to the contrast agents typicallyviastandard peptide coupling reaction between a carboxylic acid and a primary amine with a coupling reagent.The reaction can be performed in the water through a 1-ethyl-3-(3-dimethylaminopropyl) (EDC)/N-hydroxysuccinimide (NHS) reaction.The carboxyl can be activatedin situwith EDC/NHS then coupled to peptides.Thiols are not as abundant as carboxyl and amine groups in peptides.The efficient coupling between maleimide and thiol (thiol-maleimide Michael addition) makes thiol the group of choice to peptide coupling by adding thiol molecules or cysteine in the sequence [31,32].Even simpler, the DOTA like chelators such as DOTA-NHS ester and maleimide-amide-DOTA are now commercially available, making the peptide coupling progress into a one-step incubation (Fig.2B).Click chemistry is now the emerging biomarking way for site-specific radiolabeling.The most prevalent click reactions are the Cu-catalyzed azide-alkyne cycloaddition (CuAAC) and strain-promoted Diels–Alder click reaction between a tetrazine and transcyclooctene(Fig.3) [33].
Fig.2.(A) Selected macrocyclic chelators for coordinating to appropriate radionuclides.(B) Selected bifunctional DOTA analogs for peptide conjunction and radiolabeling.
Fig.3.Examples of peptide conjugation reactions: standard peptide coupling reaction; thioether bond formation between a maleimide and thiol; standard click reaction [33].Reproduced with permission 2021, The Royal Society of Chemistry.
The first procedure of peptides labeled on radionuclide is direct labeling.It is a kind of conjugation based on acyclic ligands,which exhibit rapid radiolabeling efficiency but results in a greater likelihood of decomplexationin vivo.It is utilized chiefly with nonmetallic radionuclides like18F,15O,13N and11C, the standard PET radionuclides in the early years (the 1970s) [4].In the most wellknown clinical radiotracers, 2-[18F]fluoro-2-deoxy-D-glucose ([18F]-FDG), the radionuclide18F is linked directly at a carbon position of glucose.It is now the "gold standard" in diseases diagnosis and cancer staging for the different levels of glucose consumption between lesion location and normal tissue.Thereby [18F]-FDG is taken up by more diseased cells than normal cells.The peptide and the radionuclide can be crosslinked by a chemical reaction like EDC/NHS reaction, click reaction, and thiol-maleimide Michael addition [34].Compared to the Indirect radiometal-labeling method,direct labeling is more complicated and restricted due to the characteristics of short-lived radioisotopes and rigorous labeling conditions [35].[18F] severely restrict its applications with a physical half-life of only 110 min.More importantly, the harsh radiolabeling reaction conditions (like high temperature) may lead to the inactivation and degradation of the targeting peptidein vitro.
Another widely used labeled approach requires the chelating agents or BFCs for broader applications.Stable complexes are formed through the identification of compatible chelate-metal matches.It is the primary way for radiometals labeling.Bifunctional chelators serve two purposes: securing the radiometal ion and providing a covalent link between the chelators and the targeting peptide [36].BFCs generally consist of nonfunctional analogs through the addition of functional groups for facile bioconjugate coupling.The primary goal of radiometal complexation is to form robust coordination complexes to prevent the release of free metalsin vivo.Each part is indispensable for radiometal complexation, including radiometal, ligand/chelator, linker, and bioconjugate/targeting vector [37].Stable complexes are formed through the identification of compatible chelate-metal matches.The peptide-based radiotracers can be effectively and precisely delivered to the lesion location with much less radiometal lossin vivofor imaging.The radiometals are border available for relatively milder labeling conditions and a much wider half-lives range.This facilitated their use with more vulnerable vectors, such as protein, antibodies, and peptides.
In the case of radiolabeling through a chelator, a radionuclide is complexed with the donor atoms of BFCs [29].Interactions between radiolabeling and peptide may be influenced by the traits of each other or the bifunctional chelating agent.Several issues need to be considered for the design of peptide-based radiotracers, such as solubility, low off-target toxicity, biodegradability, targeting and reaction efficiency [38,39].The peptides might be suffered from enzymatic degradation and fast renal clearance before reaching the intended targetin vivo.Most metal ion-chelate complexes are rapidly excreted with no targeting moiety due to their small/hydrophilic nature.Metal isotopes including64Cu,68Ga,177Lu,111In and90Y are mainly introduced to the peptides with the aid of chelators with polyamino polycarboxylic ligands [40].These chelators efficiently coordinate the metals with all of the amino groups and some of the carboxyls.The multiple binding sites lead to high binding strengths [41].The selection of an appropriate radiometal or ligand is critical for thein vivoexperiment.68Ga,55Co exhibited intermediate half-life, made them suitable for PET targetedimaging.They would be accumulated at target tissue and cleared relatively quickly by circulation [42].Clinical studies have shown the superior diagnostic properties of [68Ga]-DOTA-octreotate over the FDA-approved [111In]-DPTA-octreotide with better sensitivity and resolution [43].Further, the FDA approved the diagnostic use of [68Ga]-DOTA-octreotate in 2016.Radiometal-chelator complexes may have poorin vivostability because conventional bifunctional chelators such as DOTA can bind many different metal ions, leading to radiometal releasein vivo.As for64Cu chelator complexes,64Cu-TETA complexes are more stable than64Cu-DOTA [20].Another attractive alternative to DOTA, diethylenetriaminepentaacetic acid (DTPA), was widely applied for metal ions that can be chelated under mild conditions and shorter incubation times.It forms more stable radiometal-chelator complexesin vivo[44–46].An increasing number of studies have been exploring the new chelators that enable fast, stable, and radiometal complexation at the condition of near-neutral pH [47].Hetero-multivalency provides a route to increase binding affinity and enhance specificity compared with monovalent peptide ligands [48].The higher specificity can potentially serve as a unique platform to develop diagnostics and therapeutics.Aranoet al.explored the introduction of peptidic,αvβ3targeting vectors by a monodentate isonitrile donor.The monodentate nature of the targeting vector allows for the attachment of three RGD peptides onto the99mTc(CO)3core [49].This approach provided an elegant approach to enhanced targeting ability and multivalency.The corresponding complexes are stablein vivoand exhibit enhanced accumulation in target tissues compared with monovalent analogs using a polydentate chelator to attach the peptide to the metal center [50].
2.2.Radionuclide imaging for PET/SPECT imaging
Radionuclide imaging has distinctive features like high sensitivity and tomographic capability.The powerful tools employed for radionuclide diagnostics are single-photon emission computed tomography (SPECT) imaging and positron emission tomography(PET) imaging [51,52].SPECT and PET imaging modalities are the most sensitive molecular imaging techniques, providing picomolarrange sensitivity.Both methods have a limitless penetration depth and enable high sensitivity whole-body imaging of molecular targets (Table 1).Since biochemical changes generally occur before anatomical changes in disease, PET and SPECT imaging have a clear diagnostic advantage over anatomical techniques such as classical CT/MRI.The nuclides used in SPECT imaging differ from those used in PET.Radiotracers for PET or SPECT imaging emit or indirectly produce gamma rays, which were detected by PET detector gamma camera to image reconstruction of radiotracers distributionin vivo[53].
Table 1 The characteristics of representative radionuclide imaging.
The radionuclides for PET imaging are usually had short halflives such as18F (t1/2=109.8 min),64Cu (t1/2=12.7 h) [54].SPECT imaging remains the most commonly used nuclear medicine modality in the clinic.PET and SPECT imaging have a clear diagnostic advantage over anatomical techniques but lack spatial resolution [55].Since the PET or SPECT imagines have been combined with either CT or MRI, some peptides have favorable pharmacokinetic and good targeting ability.Several receptor-specific peptides and analogs have been screened, synthesized, and labeled on radioisotopes [56].Various peptide-based radionuclide probes have demonstrated good tumor uptake and nonspecific uptakes in normal organs, such as the liver and kidneys.Compared to the traditional clinical contrast agents [18F]-FDG (Fig.4A), the peptidebased radiolabeled agent may have a better imaging effect.18FFPPRGD2and showed a higher tumor-to background ratio than18F-FDG in brain lesions, but no significant difference in body lesions [57,58].The64Cu-labeled knottin peptide (targeted to integrins upregulated during angiogenesis) also displayed a higher tumor to background (normal lung) ratio compared with [18F]-FDG in the PET imaging studies [59].In this section, some specific radiolabeled peptide analogs probes were introduced: most well-known integrin targeting peptide Arg-Gly-Asp (RGD) analogs;DOTA-conjugated somatostatin (SST) receptor targeting peptide octreotide analogs (i.e., DOTATATE, DOTATOC and DOTANOC); gastrinreleasing peptide (GRP) receptor targeting peptide bombesin (BBN)fragments.
2.2.1.RGD peptide analogs
RGD peptides analogs are the most famous integrins (αvβ3) targeting vector during the past decades.The integrins (αvβ3) play a key role in angiogenesis, and angiogenesis is upregulated in various diseases, especially for different types of growing tumors, including neuroblastomas, glioblastomas, prostate, and prostate breast cancers [60].The radiolabeled RGD analogs are very abundant, including a series of cyclic RGD analogs such as c(RGDyK) (cyclo(Arg-Gly-Asp-D-Tyr-Lys)), cilengitide (cyclo(RGDf(NMe)V)).Moreover, RGD multimers have been developed for enhancing binding affinity [61].Most radiolabeled RGD peptide analogs have provided excellent tumor targeting ability and good imaging quality in animal models[62,63].Multimeric cyclic RGD peptides can improve the integrin receptor binding affinityin vitroandin vivodue to the polyvalency effect [64,65].Polyvalency profoundly affects the receptor-binding affinity andin vivokinetics [66].Liet al.developed DOTA conjugated RGD tetramer and RGD octamer with64Cu labeled PET imaging.the RGD octamer had a significantly higher integrin-binding affinity and specificity than the RGD tetramer analog,64Cu-DOTARGD octamer had higher tumor uptake and longer tumor retention than64Cu-DOTA-RGD tetramer in both tumor models [67].18F-labeled Glu-[c(RGDcK)]2([18F]FRGD2) and 2-[18F]-FPA labeled NH2-PEG3-Glu-[c(RGDyK)]2([18F]FPPRGD2) was approved by clinical investigation (Fig.4B) [68].Chenet al.found that the tetrameric RGD peptide tracer18F-FPRGD4 has significantly higher tumor uptake compared with monomeric and dimeric RGD peptide tracer analogs [69].Similarly, Caiet al.designed44Sc radiolabeled dimeric cyclic RGD peptide (cRGD)2probe chelated with DOTA for noninvasive PET imaging in the U87MG xenograft model [70].A 3-fold increase in tumor uptake of68Ga-TRAP-(RGD)3was also revealed in comparison with68Ga-NODAGA-RGD and [18F]Galacto-RGD [71].Similar works were reported by labeling various radiometals, such as64Cu,68Ga and99mTc [72].18F can be directly radiolabeled to the RGD peptide analogs such as [18F-galacto]-c(RGDfK) and18F-AH111585 [60,73].By introducing a linker SAA (7-amino-L–glycero-L-galacto-2,6-anhydro-7-deoxyheptanoic acid) through the side chain of the lysine group from c(RGDfK), the [18F-galacto]-c(RGDfK) was synthesized.The core peptide sequence of18FAH111585 is RGD4C, cyclizedviadisulfide bonds.AH111585 consisted of cyclic RGD peptide and flexible linker poly(ethylene glycol) to stabilize the radiolabeled peptide.These two18F-labeled RGD analogs were now the candidates of radiopharmaceutical for clinical application.Besides18F, some radiometals can also be directly labeled to the peptide, such as64Cu,68Ga,177Lu,111In, and90Y.[123I]-cyclo(RGDyV) was the first radiolabeled peptide analogs evaluated preclinically in detail.Bifunctional chelators(DTPA, DOTA, NOTA, TETA, and TE2A) are widely applied for efficiency delivering and targetingin vivo[74].For identifying the effects of bifunctional chelators on the radiopharmaceuticals, Yooet al.investigated five different64Cu-labeled chelators conjugated with the same RGD peptide c(RGDyK) by PET imaging (Fig.4C)[75].These probes with different bifunctional chelators show appreciable differences in tissue uptake and clearance.They found propylene cross-bridged chelators exhibited higher tumor uptake and rapid body clearance.
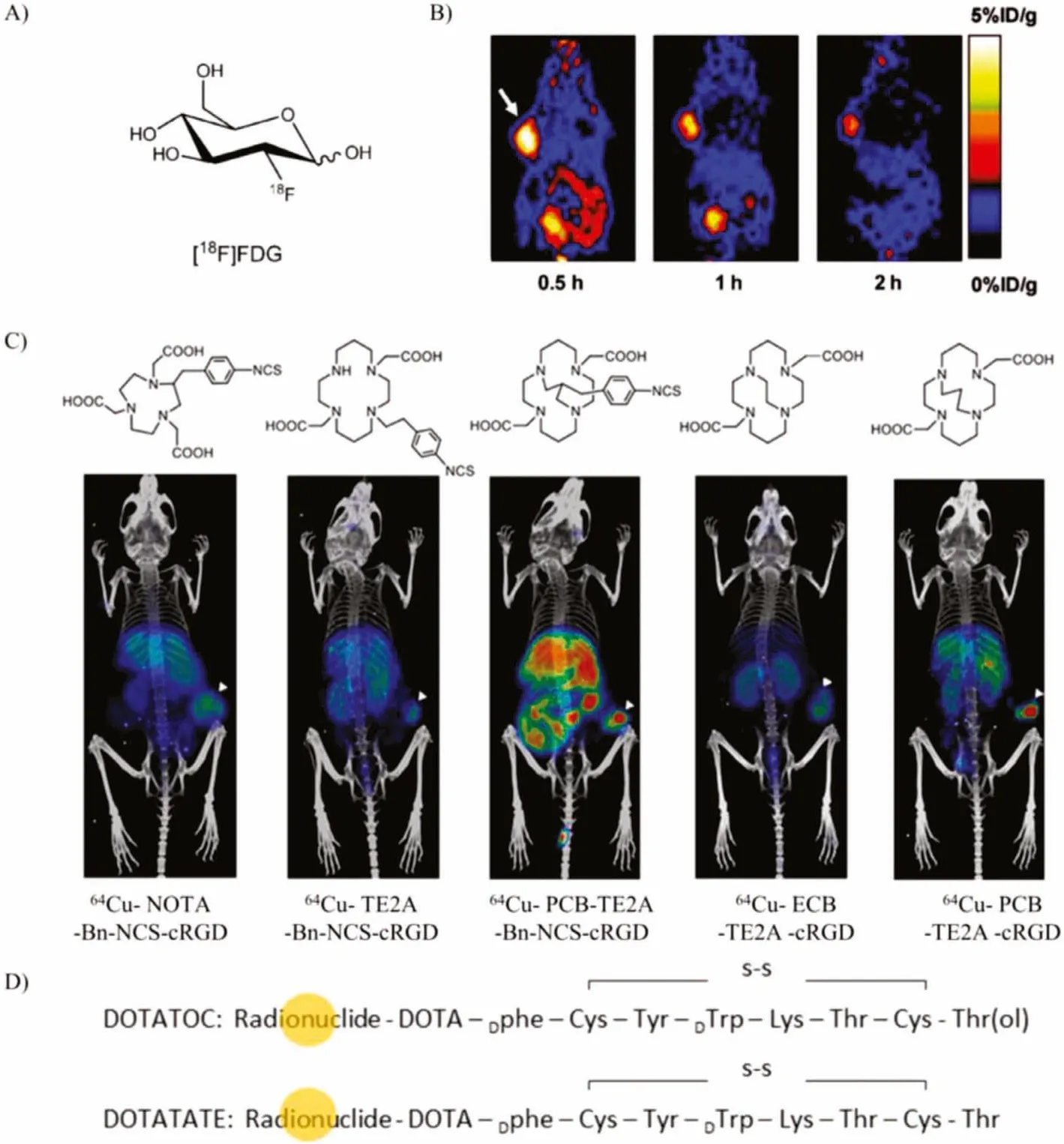
Fig.4.(A) The structure of [18F]-FDG.(B) The PET images of U87MG tumor-bearing mice after injection of [18F]FPRGD2.Copied with permission [68].Copyright 2007,Springer-Verlag.(C) The PET imaging of 64Cu with different radiolabeled BFC-c(RGDyK) conjugates.White arrowhead indicates tumor.Copied with permission [75].Copyright 2018, American Chemical Society.(D) The structure of radionuclide labeled DOTA-somatostatin analogs for various clinical applications.
2.2.2.Octreotide analogs
One of the most widely investigated peptide analogs is the octreotide analog.Octreotide is a cyclic octapeptide (DPhe1–Cys2-Phe3-DTrp4-Lys5-Thr6-Cys7-Thr8-ol).It was derived from somatostatin analogs with a high affinity to somatostatin receptors(SSTrs).SSTrs are overexpressed on most tumors, such as neuroendocrine tumors, gliomas, breast cancer, and small cell lung cancers (SCLC) [76].Since that SSTrs represent one of the best examples of targets for radiolabeled peptide probesin vivo[77].Several somatostatin analog peptides were developed to improvein vivostability and extend the application of targeting.[111In-DTPA]-octreotide (OctreoScan) was the first FDA-approved peptide-based radiopharmaceuticals for neuroendocrine tumors (approveal at 1994).Octreotide analogs (TOC), Tyr3-octreotide (TATE), and NaI3-octreotide (NOC) (Fig.4D), these octreotide analogs were mostly labeled with DOTA chelator to form thermodynamically and kinetically stable complexes with radiometals well-known for DOTATATE,DOTATOC, and DOTANOC.In many works, the radiometals like44Sc,55Co,58mCo,64Cu,99mTc,68Ga,111In,90Y,86Y,152Tb,177Lu,213Bi and225Ac were labeled with DOTATATE, DOTATOC, or DOTANOC observed the PET/SPECT imaging effect under different labeling conditions, chelator agents, and targeting vector [78–84].The experimental results indicated that accessing radiometals or BFCs or even different targeting analogs might greatly influence thein vivoperformance and metabolism.[111In/90Y] DOTANOC showed higher affinity to sstr3 and sstr5 when compared to [111In/90Y]-DOTATOC [85].SST receptor targeting has recently changed focus from SST agonists to antagonists.111In-DOTA-BASS (111In-DO3ApNO2-Phe-c(DCys-Tyr-DTrp-Lys-Thr-Cys)-DTyr-NH2) is the first clinical SST antagonist applied as a tracer [86].It was reported that111In-DOTA-BASS exhibited higher tumor uptake and improved visualization of metastatic neuroendocrine tumors compared to clinical [111In-DTPA]-octreotide.Radiopharmaceutical agents111In pentetreotide,99mTc depreotide,68Ga-DOTATATE,68Ga-DOTATOC and64Cu-DOTATATE have been approved as radiopharmaceutical agents to detect tumors [87].Furthermore, the177Lu-DOTATATE for the treatment of somatostatin receptor-positive gastroenteropancreatic tumors was approved recently [88].
2.2.3.α-Melanocyte stimulating hormone analogs
α-Melanocyte stimulating hormone (α-MSH), a linear 13 amino acid peptide (SYSMEHFRWGKPV), mainly regulates skin pigmentation.Theα-MSH receptors are distributed in more than 80%of human melanoma metastases.α-MSH peptides are an attractive targeting candidate for melanoma targeting.Variousα-MSH peptides radiolabeled with18F,99mTc,111In,125I,67Ga,86Y and64Cu [89].Severalα-MSH analogs with better targetingin vivolike(Arg11)CCMSH were designed [90].Subsequently, DOTA-conjugated and remediated CCMSH analogues, including [DOTA]-ReCCMSH and[DOTA-Arg11]-ReCCMSH were developed [91,92].Renet al.found that transition metal rhenium-cyclizedα-MSH peptide isomer Ac-D-Lys-ReCCMSH (Arg11) had specificin vivotargeting than18Flabeled linearα-MSH peptide for better tumor uptake and retention [93].As for melanoma staging and metastases identification, the widest clinic applied [18F]FDG was not a practical and specific contrast agent for glucose is not the primary energy source for melanomas [94].Chenget al.first developed an18F radiolabeledα-MSH analog to imaging the MC1R [95].This18Flabeled NAPamide revealed a stark difference in tumor accumulation between the B16/F10 model (1.19%±0.11% ID/g) and A375M model (0.46%±0.11% ID/g) model as a result for high and low MC1R expression, respectively.Guoet al.proved that67Ga-DOTAGlyGlu-CycMSH exhibited favorable melanoma targeting and imaging properties as SPECT imaging probes [96].68Ga-DOTA-GGNle-CycMSHhex was designed to target to melanocortin-1 receptor(MC1R) for PET imaging.Yanget al.firstly evaluated the possibility of68Ga-DOTA-GGNle-CycMSHhex in patients with advanced-stage melanoma [97].The melanoma imaging in patients demonstrated the clinical relevance of MC1R, highlighting the potential of68Ga-DOTA-GGNle-CycMSHhex as an MC1R-targeting melanoma imaging probe for melanoma metastases in the brain, as well as in lung,connective tissue, and small intestine of patients.MC1R is a valid molecular target for melanoma during early development and is present in advanced stages of melanoma and later generations of daughter metastases.
2.2.4.Bombesin (BBN) analogs
Bombesin (BBN) is a 14-amino acid peptide with high affinity and specificity to the gastrin-releasing peptide receptors (GRPs).Overexpression of GRPrs has been demonstrated in various cancers, including small cell lung cancer (SCLC), prostate, breast, pancreas, and gastrointestinal tumors [98,99].The application of BBN peptide-based probes is more relevant clinically than that of SST peptides due to the broad spectrum of BBN specificity for various tumors.Many radioisotope probes based on BBN analogues’critical amino acid sequence, BBN7–14, were developed.The radioisotopes were mainly incorporated at the N-terminus of the peptide.Besides using single peptide ligands, multivalent peptide dimers and tetramers or heterodimers for dual receptor-targeting can be utilized for improved binding affinity.A BBN-RGD heterodimer was synthesized by coupling two targeting peptides with a flexible linker glutamic acid [100,101].Liuet al.reported a PET peptides probe of64Cu labeled RGD-bombesin heterodimer for dual integrin- and GRPR-targeted imaging [102].For the integrin-and GRPR-positive tumors PC-3,64Cu-NOTA-RGD-bombesin showed the advantages over the sum of64Cu-NOTA-RGD and64Cu-NOTAbombesin.Moreover, in the only integrin-positive 4T1 model, the accumulation of64Cu-NOTA-RGD-bombesin at tumor sit was still higher than 64Cu-NOTA-RGD.However, the PET tumor imaging of18F labeled RGD-bombesin is better than64Cu within 2 h after injection.64Cu-NOTA-RGD-bombesin showed much higher and persistent imaging signals from 4 h to 20 h for the short halflife of18F.This made64Cu-NOTA-RGD-bombesin more available for GRPR-positive tumor-targeted therapy.Dual targeting of integrin and GRPr significantly improved tumor targeting efficacy and pharmacokinetics with various radioisotopes, such as18F,64Cu and68Ga, compared with RGD and BBN analogs in prostate and breast cancer models.
2.2.5.Other peptide analogs
Cystine knot peptides, also known as knottins, are small(~4 kDa) peptides with a stable core motif formed by multiple disulfide bonds.Knottins are well suited forin vivotumor-targeting applications for their nonimmunogenic, high thermal, proteolytic stability, and resistance against proteolytic degradation.Knottins can be chemically modified to tailor theirin vivopharmacokinetic properties.The integrin family is a frequent target of knottins.The integrin family, mainly,αvβ3,αvβ5, andαvβ6are significantly upregulated on tumor neovasculature and many tumor cells.Hackelet al.found that18F radiolabeled cystine knot peptides showed promise for molecular imaging of integrinαvβ6overexpression in pancreatic adenocarcinoma xenografts model [103].Jianget al.evaluated the ability to image integrinαvβ3–targeting PET probe64Cu-NOTA-3–4A in a mouse model for carotid atherosclerotic plaques [104].The results showed that64Cu-NOTA-3–4A might have clinical utility as a PET probe for atherosclerosis imaging or evaluating therapies used to treat atherosclerosis [105].Moreover,this kind of64Cu-DOTA-Knottin Peptide probe can also be applied in PET for imagingαvβ3–targeting PET probe of Tumor Neovascularization.
Metal nanoclusters have attracted considerable attention because of their unique properties of ultrasmall size, good biocompatibility, and high stability [106,107].64Cu as a PET imaging agent generally has been introduced into biomaterials through macrocyclic chelators, which may lead to the misinterpretation of PET imaging results due to the detachment of64Cu.In order to improve radiolabeling stability and ensure diagnostic accuracy, Gaoet al.developed ultrasmall chelator-free [64Cu]CuNC@BSALHRH nanoclusters for PET imaging in the orthotopic lung cancer model [108].The bovine serum albumin (BSA) was used as a scaffold to coordinate with the64Cu.Tumor target peptide luteinizing hormone-releasing hormone (LHRH) was conjugated to BSA molecules to prepare [64Cu]CuNC@BSA-LHRH.The nanocluster [64Cu]CuNC@BSA-LHRH showed four times higher tumor uptake than [64Cu]CuNC@BSA.The PET imaging using [64Cu]Cu nanoclusters as tracers showed more sensitive, accurate, and deep penetration imaging of orthotopic lung cancerin vivothan near-infrared fluorescence imaging.The direct incorporation and chelator-free radiolabeling strategy like64Cu-loaded liposomes provide new biomedical research tools for PET molecular imaging.
3.Combined imaging
Since each imaging system has its limitations and advantages,multiple imaging modalities can obtain more information in less time [109].Two or more modalities are used to compensate for the weaknesses and exploit individual strengths.
CT and MRI imaging are the most commonly used imaging techniques for non-invasiveness and high spatial resolution [55].As for CT imaging, the X-ray absorption between cancerous lesions and their surrounding normal tissues is different [110].The different X-ray absorption created the high-contrast CT imaging with detailed morphological information [111].CT imaging has proven invaluable in identifying and assessing tumors in the clinic.It is truly the workhorse for modern-day clinical imaging.Although CT imaging can provide cross-sectional images at high resolution, especially for bones, organs, and tissues, it is limited in differentiating soft tissues with similar densities [106].In vivo, MRI is a highly versatile, non-invasive, and non-destructive imaging tool for characterizing disease.This imaging modality involved the detection of the signal from hydrogen nuclei in water [112].MRI signal intensity depends on the density of water in soft tissue and has been well developed for visualizing the body’s internal structure and soft tissue morphology [113].MRI has excellent clinical utility and several significant advantages, including no need for ionizing radiation, unlimited depth of penetration, high spatial resolution, and soft-tissue contrast [114].A significant limitation of MRI is its low sensitivity [115].Both CT and MRI may need a high dose of contrast.These large amounts of imaging agents are many log orders higher than that needed for PET or SPECT.Although SPECT and PET imaging has a nanomolar range sensitivity, they have a fundamental shortcoming, the limited spatial resolution [116].However, CT imaging and MRI imaging had the advantages of high spatial resolution penetration.Recently, SPECT and PET imaging, combined with the high spatial resolution of CT/MRI images, play an increasingly important role in clinical application.
Dual-modality imaging has become a gold standard in clinical oncology because it can provide both molecular and anatomical information.The inorganic nanoparticles can be easily decorated with different targeting ligands.Radionuclide-label peptide nanoparticulate contrast agents are one of the most applied multimodality imaging agents for SPECT/CT imaging or PET/CT imaging[117,118].Sangeeta N.Bhatiaet al.designed protease-responsive imaging sensors for malignancy (PRISM) that can be employed to assess disease progression (Fig.5A).The tumor-targeting peptide, protease substrate, and imaging tracers were displayed on the nanosensor’s surface to target cancer cells in the acidic tumor microenvironment (TME).Higher tumor-specific PET imaging signals were observed.It has been reported that 3.11±0.23-fold tracer accumulation in the tumor-bearing lungs exposed to the targeting64Cu-PRISM relative to the control sensor (Figs.5B and C) [119].The localization capability arose from acidosis-targeting and loading with64Cu.PET-CT imaging used64Cu-loaded PRISM to localize CRC lung tumors and reveal tumor-specific sensor accumulation.Kimuraet al.developed18F and68Ga radiolabeled cystine knot PET/CT tracers for effectively detecting multiple cancers in different regions and fibrotic changes in the lungs of patients [120].In pancreatic, cervical and lung cancer, the knottin PET/CT tracers demonstrated rapid and sustained accumulation in diseased tissues with relatively low background uptake in healthy organs or regions of the body prone to different cancers or fibrosis (lung and liver).These results suggest that radiolabeled cystine knot PET/CT tracers had broad clinical application in diagnosing [121].
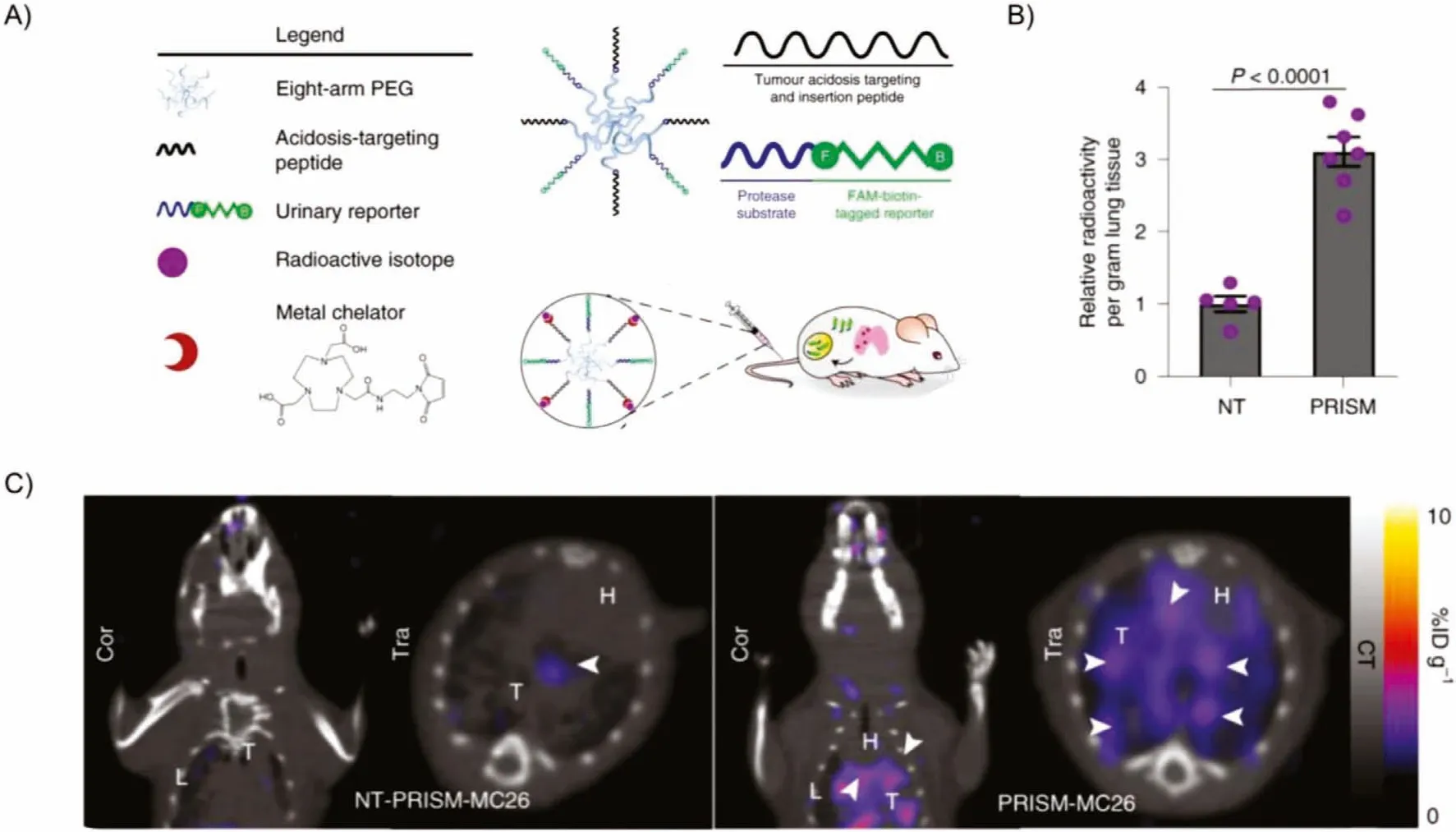
Fig.5.(A) The functional moieties of PRISM for tail intravenous injection.(B) Ex vivo quantification of the radioactivity accumulated in the lungs extracted from animals imaged with 64Cu-non-targeting (NT)-PRISM or 64Cu-PRISM.(C) Representative PET-CT images of the lung in animals imaged with 64Cu-non-targeting (NT)-PRISM or 64Cu-PRISM.Reproduced with permission [119].Copyright 2021, Springer Nature.
Iodinated contrast agents could elegantly combine the advantages of SPECT/PET (ultra-high sensitivity) and CT/MRI (location of organs/tissues), providing a better diagnosis of various diseases.Sunet al.desined a125I-(cRGD)2-iron oxide nanoparticles(IONPs) nanoprobe which could be successfully used as T1-T2-MRI/SPECT contrast agents for high-resolution and high-sensitivity of tumor imagingin vivo[122].Guet al.reported a radiolabeled peptide probe for SPECT/CT dual-modality imaging.This probe was based on poly(ethylene glycol)-b-poly(lthyroxine) (PEG-PThy)block copolypeptide.The high SPECT/CT signals were observed at the tumor site for a long time and possess highin vivostability.At 8 h postinjection,125I-cRGD-PThyN revealed a boosted tumor accumulation of around 2.3 times higher than125I-PThyN (Fig.6A) [123].Compared to clinically used iohexol cRGD-functionalized PThyN (cRGD-PThyN) exhibits largely enhanced CT contrast in both subcutaneous B16F10 melanoma (>13-fold) and orthotopic A549 lung cancer.Multifunctional and water-soluble superparamagnetic iron oxide (SPIO) nanocarriers were developed for targeted drug delivery and positron emission tomography/ magnetic resonance imaging (PET/MRI).Yanget al.conjugated cyclo(Arg-Gly-Asp-D-Phe-Cys) (c(RGDfC)) peptides and64Cu onto the PEGylated SPIO [124].According to PET/MRI imaging results, cRGDconjugated SPIO nanocarriers also had a much higher tumor accumulation level than cRGD-free SPIO nanocarriers; meanwhile,cRGD-conjugated SPIO nanocarriers had a similar MRI r2relaxivity to that of an FDA approved SPIO-based MRI contrast agent,Feridex.Besides traditional iron oxide nanoparticles, gold nanoparticles have also been extensively explored for cancer diagnosis.The chemokine receptor 5 (CCR5) has emerged as a promising prognostic biomarker for triple-negative breast cancer (TNBC).Zhaoet al.firstly reported Au nanoparticles doped with199Au atoms for SPECT imaging.D-Ala1-peptide (D-A1STTTNYT-NH2) was conjugated to the nanoparticles for CCR5 targeted.The SPECT/CT results showed that the199Au-AuNP-DAPTA was evenly distributed in the TNBC tumors, improved biodistribution profile, and significant enhancement in tumor SPECT/CT imaging sensitivity and specificity (Fig.6B) [125].Gutierrezet al.prepared a kind of177Lu labeled AuNPs conjugated to c[RGDfK(C)] for SPECT/CT images toαvβ3integrin-positive U87MG tumors in mice.The results showed that177Lu-AuNP-c[RGDfK(C)] delivered the highest tumor radionuclide-absorbed dose compared to177Lu-AuNP-c[RGDfK(C)],177Lu-AuNP, or177Lu-RGD [126].177Lu-AuNP-c[RGDfK(C)] demonstrated properties suitable for targeted radionuclide therapy of tumors expressingαvβ3integrins.Nanoclusters are ultra-small clusters.Gaoet al.developed targeting peptide-coated Au clusters for precisely assessing the risk of primary tumor invasion/metastasis[127].With intrinsic red fluorescence and a specific mass signal,these clusters detect the MT1-MMP expression level in primary human lung carcinoma and human renal carcinoma tissue sections by Au-CL-based 2D-LA-mass mapping method.The assessment accuracy of this pathologic method was verified by CT/MRI molecular imaging of cancer patients and traditional hematoxylin.The imaging results showed that early diagnosis of primary tumor invasion/metastasis could be achieved by 2D-LA-mass mapping of MT1-MMP in tumor tissues.This method would have significant benefits for clinical therapy and the prognosis of primary tumor patients.
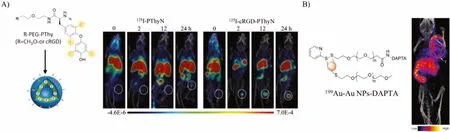
Fig.6.(A) The structure of 125I-PThyN and 125I-cRGD-PThyN and the related in vivo SPECT/CT imaging.Reproduced with permission [123].Copyright 2019, Wiley Publishing Group.(B) Nano SPECT/CT image after injection of 199Au-AuNP-DAPTA probe.Copied with permission [125].Copyright 2016, Wiley Publishing Group.
Besides MRI/CT, many other imaging modalities can also be combined with PET/SPECT.Zhaoet al.developed a novel combination probe of NIR-II/PA/SPECT imaging:125I-labeled QT-RGD[128].This probe was constructed by a NIR-II emissive organic fluorophore and two cyclic-(arginine-glycine-aspartic) (cRGD) peptides and labed by125I.These probe exhibited great tumor targeting capability and emitted intensive NIR-II/photoacoustic (PA)/(SPECT)signals, allowing specific and sensitive multimodal visualization of tumorsin vivo.Yinet al.reported a125I labeled matrix metalloproteinase-2 (MMP-2)-activatable peptide probe to detect metastatic lymph nodes (MLNs) accurately [129].This peptide probe was constructed with an MMP-2-cleavable peptide sequence KCPLGVRGY and a tumor-targeting peptide cRGD labeled with a near-infrared dye (Cy5), a quencher (QSY21), and a radionuclide(125I).In vivoimaging studies have demonstrated the outstanding ability of this NIRF/SPECT dual-modal imaging probe for its excellent tumor-targeting specificity and imaging sensitivity enabled.
Dual-modality imaging systems have been rapidly developed to provide more information and combine the advantages of two kinds of imaging technique.They may help the clinical applications to be more sensitive and accurate.
4.Conclusion
Targeted imaging techniques at present play a fundamental role in modern diagnostics because they provide accurate and specific disease information.Specific peptide receptors overexpressed in many diseases attract enormous interest in developing specific targeting peptides.Since conventional nonspecific contrast agents suffer from low targeting efficiency, radiolabeled peptide probes have emerged as an essential targeted molecular imaging agent class.This article introduced some peptide-based radiocontrast agents and discussed many typical radiolabeled peptide probes for tumor imaging concerning their design strategies and applications in PET/SPECT imaging.
There are no depth penetration limits of PET and SPECT for the high-energy gamma-ray photons emitted by ultra-low-dose radiocontrast agent.Moreover, PET and SPECT are more sensitive than other imaging modalities.However, PET and SPECT offers lower spatial resolution so they often combine with other imaging systems such as MRI and CT to provide spatial resolution anatomical information.Radiolabeled peptide probes have become significant imaging agents for tumor imaging.An ideal peptide conjugated radiocontrast agent should possess favorable pharmacokinetic properties (rapid distribution and high metabolic clearance) to produce a sufficient "target-to-background" ratio of imaging signals.Compared to other radiolabeled probes, peptides probes offer clear advantages for the specific targeting ability, high target/background ratio, excellent tumor target and desirable pharmacokinetics, which made radiolabeled peptide probes widely applied in tumor imaging.Despite the significant progress in developing peptide contrast agents for tumor imaging, these kinds of peptides probes may have a shortin vivobiological half-life.Chemical modifications were needed to enhance their practical properties, and their application in preclinical and clinical studies was still in the early stages.More sensitive, specific, and stable radiolabeled peptide probes are expected to develop.The development and application of radiolabeled peptide probes have a long way to go.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Acknowledgment
This work was supported by the National Key R&D Program of China (No.2018YFE0205400).
 Chinese Chemical Letters2022年7期
Chinese Chemical Letters2022年7期
- Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
- Recent development in selective Tau tracers for PET imaging in the brain
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging